Bild : „334 “ von Evan Blaser. License: CC BY-SA 2.0
- Wichtige Definitionen zum Verständnis der Pharmakodynamik
- Rezeptoren
- Effektoren
- Affinität
- Selektivität
- Spezifität
- Agonismus
- Antagonismus
- Synergismus
- Wirksamkeit
- Potenz
- Exkretion
- Elimination
- Rezeptoren und Effektoren
- Rezeptoren
- Effektoren
- Hochregulierung und Herunterregulierung
- Hochregulierung
- Downregulation
- Dosis-Wirkungs-Kurven
- Bindungskurven
- Rezeptoren ersetzen
- Toxizität, Toxizitätskurven und therapeutisches Verhältnis
Wichtige Definitionen zum Verständnis der Pharmakodynamik
Rezeptoren
Ein Rezeptor ist die Komponente (Makromolekül) einer Zelle (auf oder innerhalb der Zelle), die mit dem Medikament interagiert, und diese Interaktion führt zu einer Kette von Ereignissen, die die Aktivität der Zelle verändern.
Effektoren
Effektoren sind Moleküle, die als Reaktion auf den Wirkstoff (oder genauer gesagt auf den Wirkstoff-Rezeptor-Komplex) wirken und an der oben genannten Kette intrazellulärer Ereignisse teilnehmen, die zu den Wirkungen des Wirkstoffs führen.
Affinität
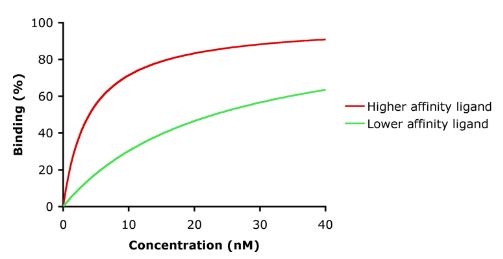
Affinität ist das Maß für die Stärke der Bindung zwischen der Droge und ihrem Rezeptor. Die Affinität eines Arzneimittels zu seinem Rezeptor hilft bei der Bestimmung der Arzneimitteldosis: Eine niedrige Affinität würde darauf hinweisen, dass eine höhere Dosis erforderlich ist, um genügend Arzneimittel-Rezeptor-Komplexe zu bilden, die zu einer signifikanten Wirkung führen (siehe Abbildung).
Selektivität
Selektivität ist die Vorliebe des Arzneimittels für einen Rezeptor oder seinen Subtyp (im Vergleich zu anderen Rezeptoren oder anderen Subtypen). Mit anderen Worten, es ist die Fähigkeit des Arzneimittels, eine bestimmte pharmakologische Wirkung gegenüber anderen zu erzielen. Wenn ein Arzneimittel selektiv ist, bindet es vorzugsweise an einen Rezeptor, kann aber durch Erhöhung seiner Konzentration auch an andere Rezeptoren binden. So blockiert Verapamil normalerweise die Ca-Kanäle, kann aber in hohen Konzentrationen auch die Na-Kanäle blockieren.
Spezifität
Spezifität ist die Fähigkeit eines Arzneimittels, nur an einen Rezeptor zu binden. Atropin ist technisch gesehen spezifisch, da es nur an Acetylcholin (ACh)-Rezeptoren wirkt. Atropin ist jedoch nicht selektiv, da es an alle Subtypen bindet und zahlreiche pharmakologische Wirkungen hervorruft.
Agonismus
Wenn das Medikament mit dem Rezeptor interagiert, um eine Reihe von Ereignissen hervorzurufen, die zu einer pharmakologischen Wirkung führen, wird dies als Agonismus bezeichnet. Zum Beispiel ist Isoprenalin ein β-Adrenorezeptor-Agonist.
Antagonismus
Wenn das Medikament mit dem Rezeptor interagiert, aber keine Reihe von Ereignissen hervorruft und dadurch die potenzielle Wirkung (physikalisch oder chemisch) „blockiert“, spricht man von Antagonismus. Zum Beispiel ist Propranolol ein β-Adrenorezeptor-Antagonist.
Antagonisten können kompetitiv (durch reversible Bindung an den Rezeptor) oder nicht-kompetitiv (durch irreversible Bindung an den Rezeptor oder durch allosterische Veränderung der Konformation des Proteins/Enzyms) sein. Die Wirkung eines kompetitiven Hemmstoffs kann durch Erhöhung der Konzentration des Agonisten umgekehrt werden, während dies bei nicht-kompetitiven Hemmstoffen nicht der Fall ist.
Synergismus
Wenn die Wirkung eines Arzneimittels verstärkt wird, wenn es zusammen mit einem anderen Arzneimittel verabreicht wird, spricht man von Synergismus. Beispiele sind Aspirin und Paracetamol.
Wirksamkeit
Wirksamkeit ist die Eigenschaft eines Arzneimittels, die (gewünschte) biologische Wirkung zu erzielen. Die Potenz ist die Konzentration eines Arzneimittels, die erforderlich ist, um eine Wirkung mit einer bestimmten Intensität zu erzielen.
Potenz
Sie wird in der Regel als die Konzentration (oder Dosis) berechnet, die erforderlich ist, um 50 % der maximalen Wirkung des Arzneimittels zu erzielen (EC>50).
Bei der Anwendung dieser Konzepte im klinischen Bereich gibt die Potenz lediglich die Dosis des Arzneimittels an, während die Wirksamkeit das Ausmaß der Reaktion (unabhängig von der Dosis) angibt.
Exkretion
Die Exkretion ist die irreversible Entfernung des Arzneimittels aus dem Körper. Die Ausscheidung erfolgt hauptsächlich über die Leber und die Nieren, aber auch andere Organe (z. B. die Lunge) können beteiligt sein.
Elimination
Elimination ist der Prozess der Inaktivierung der Droge mit/ohne tatsächliche Entfernung aus dem Körper. Die Eliminierung kann durch Ausscheidung oder Metabolisierung/Biotransformation des Arzneimittels erfolgen. Während es sich bei der Elimination also um die bloße Inaktivierung des Arzneimittels handelt, ist die Exkretion der physikalische Transfer des Arzneimittels (in aktiver oder inaktiver Form) aus dem Blutkreislauf in die Ausscheidungsflüssigkeiten, wie Urin und Galle.
Rezeptoren und Effektoren
Rezeptoren
Es gibt fünf Grundtypen von Transmembranrezeptoren, die nach Bindung an die Droge oder den Liganden auf unterschiedliche Weise wirken (siehe Abbildungen).
![]()
- Intrazelluläre Rezeptoren: Hierbei handelt es sich um Proteinrezeptoren, bei denen der Wirkstoff die Plasmamembran passieren muss; daher muss der Wirkstoff lipophil sein. Steroide zum Beispiel wirken über diesen Mechanismus.
- Transmembranenzyme: Ein Medikament bindet an die extrazelluläre Komponente dieses Rezeptors, was eine enzymatische Reaktion in der intrazellulären Komponente auslöst.
- Tyrosinkinase: Wenn ein Medikament an die extrazelluläre Komponente dieses Rezeptors bindet, führt dies intrazellulär zu einer Dimerisierung der beiden Teile des Rezeptors. Diese Dimerisierung aktiviert die Tyrosinkinase-Enzyme, was zu einer Phosphorylierung von Tyrosinmolekülen an Zielproteinen führt. Wachstumshormone und Interferone wirken über JAK-STAT-Kinaserezeptoren.
- Ligandengesteuerte Ionenkanäle: Diese Ionenkanäle sind ligandengesteuert, d.h. sie sind geschlossen, bis der Rezeptor an das Medikament bindet, das dann bestimmte Ionen passieren lässt. So bewirken beispielsweise Medikamente, die GABA-Rezeptoren an den Neuronen stimulieren, einen Chlorideinstrom (der zu einer Hyperpolarisierung und damit zu einer Hemmung führt).
- G-Protein-gekoppelte Rezeptoren: Ähnlich wie bei Tyrosinkinase-Rezeptoren führt die Bindung des Arzneimittels an den Rezeptor zur Interaktion des G-Proteins mit dem Rezeptor. Dieses aktivierte G-Protein führt dann zu der gewünschten pharmakologischen Reaktion durch ein oder eine Reihe von Effektormolekülen oder Second Messengers. G-Protein-gekoppelte Rezeptoren sind weit verbreitete Rezeptortypen im Körper.
Effektoren
Signaltransduktion
Wenn ein Medikament an den Rezeptor bindet (extrazellulär), wirkt es als „Signal“ für das nachfolgende Ereignis oder die nachfolgenden Ereignisse (intrazellulär), das/die schließlich zur pharmakologischen Reaktion führt/führen. Diese Übertragung des Signals wird als Signaltransduktion bezeichnet, und die intrazelluläre Ereigniskette, die an diesem Prozess beteiligt ist, wird als Signaltransduktionskaskade bezeichnet.
An dieser Ereigniskette kann eine Reihe von Molekülen (so genannte Second Messenger) beteiligt sein. Die Funktion der Kaskaden besteht darin, das Signal des Wirkstoffs zu verstärken. Wie bereits erwähnt, wirken G-Protein-gekoppelte Rezeptoren über second messengers. Ein G-Protein enthält eine α-Untereinheit, die Guanosintriphosphat (GTP) bindet, sowie die β- und γ-Untereinheiten, die das Protein in der Membran verankern.
- Die α-Untereinheit dissoziiert nach der Bindung des Wirkstoffs an den Rezeptor mit den beiden anderen, und die beiden dissoziierten Komplexe können an andere Enzyme binden, um die Kaskaden zu erzeugen.
- Ein üblicher Mechanismus ist die Bindung der α-Untereinheit an das Adenylylzyklase-Enzym und dessen Aktivierung, wodurch ATP in der Zelle in zyklisches AMP (cAMP) umgewandelt wird. (Die Aktivierung wird durch den Gs-Typ des G-Proteins verursacht; Gi hemmt die Adenylylzyklase). Die erhöhten anorganischen Phosphatkonzentrationen können durch Phosphorylierung von Proteinen an das Ziel- oder andere intermediäre Effektormoleküle binden.
Andere Beispiele für Second Messenger sind Diacylglycerin (DAG) und Inositol-1,4,5-Triphosphat (IP3), die beide durch das Gq-Typ G-Protein aktiviert werden; da G-Protein-Rezeptoren die Phosphorylierung als Wirkmechanismus nutzen, sind sie oft anfällig für das Phänomen der Desensibilisierung.
Hochregulierung und Herunterregulierung
Hochregulierung
Hochregulierung (d.h., Erhöhung der Anzahl) von Rezeptoren tritt auf, wenn die Aktivität des Rezeptors geringer ist als üblich (z. B. durch die langfristige Verabreichung eines Antagonisten). Beispielsweise werden durch die Verabreichung von Betablockern die β-Adrenorezeptoren hochreguliert. Wenn β-Blocker abrupt abgesetzt werden, kann dies aufgrund der plötzlichen Stimulierung einer großen Anzahl von β-Adrenorezeptoren zu einer Rebound-Hypertonie führen.
Downregulation
Downregulation (d. h. Abnahme der Anzahl) ist das Gegenteil von Upregulation. Sie tritt bei wiederholter oder langfristiger Verabreichung eines Agonisten auf. Neben der Downregulation kann es auch zu einer Desensibilisierung des Rezeptors gegenüber dem Arzneimittel kommen. Dabei handelt es sich um eine physikalisch-chemische Veränderung des Rezeptors, die ihn für den Wirkstoff unempfindlich macht; dies wird auch als Tachyphylaxie bezeichnet und tritt z. B. bei chronischem Drogenkonsum auf.
Dosis-Wirkungs-Kurven
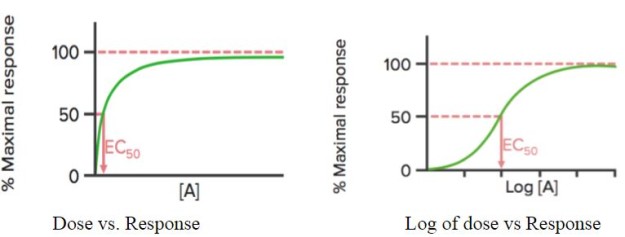
Wenn die Wirkung des Medikaments gegen seine Dosis aufgetragen wird, ergibt sich eine hyperbolische Kurve; wird der Logarithmus der Dosis (log) verwendet, ergibt sich eine sigmoidale Kurve.
Mit Hilfe dieser Kurve lässt sich die wirksame Konzentration (oder Dosis) des Arzneimittels ermitteln, bei der 50 % der maximalen Reaktion (EC50) des Arzneimittels erreicht wird. Wie bereits erwähnt, wird EC50 zur Berechnung/Vergleich der Wirksamkeit von Arzneimitteln verwendet.
Emax ist die (Mindest-)Konzentration, bei der die maximale Wirkung des Arzneimittels beobachtet wird. Bei Emax sind alle Rezeptoren durch das Medikament besetzt.
Bindungskurven
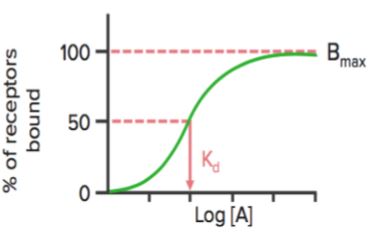
Wenn der Rezeptorbindungsstatus (% der gebundenen Rezeptoren) gegen die Medikamentenkonzentration (oder log) aufgetragen wird, zeigt sich eine ähnliche hyperbolische (oder sigmoidale) Kurve.
Anhand dieses Diagramms können wir die effektive Konzentration berechnen, bei der 50 % der Rezeptoren gebunden sind (Kd). Kd zeigt die Bindungsaffinität des Arzneimittels an – ein hoher Wert von Kd bedeutet, dass eine hohe Konzentration des Arzneimittels erforderlich ist, um 50 % der Rezeptoren zu binden, was auf eine niedrige Affinität hinweist.
Bmax ist die maximale Bindung des Arzneimittels. Es ist die Konzentration, ausgedrückt in Pikomol pro mg Protein, bei der alle spezifischen Rezeptoren an den Wirkstoff gebunden sind. Daher kann Bmax verwendet werden, um die Dichte der Rezeptorstelle in einem bestimmten Präparat zu berechnen.
Rezeptoren ersetzen
Wenn ein Agonist an einen Rezeptor bindet, kann er eine Reihe von Ereignissen hervorrufen, die schließlich zu der pharmakologischen Reaktion führen. Diese Reihe von Ereignissen kann die Wirkung des Arzneimittels so verstärken, dass nur eine relativ geringe Menge der Arzneimittel-Rezeptor-Komplexe erforderlich ist, um eine maximale Reaktion zu erzielen. In diesem Fall bleibt bei Erreichen der maximalen Reaktion ein bestimmter Prozentsatz der Rezeptoren von der Droge unbesetzt. Diese Rezeptoren werden als Ersatzrezeptoren bezeichnet.
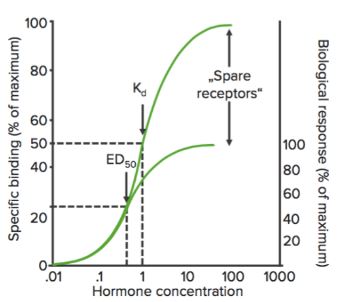
Ersatzrezeptoren erhöhen zumindest theoretisch die Empfindlichkeit des Medikaments.
Toxizität, Toxizitätskurven und therapeutisches Verhältnis
Nachteilige Wirkungen eines Medikaments sind normalerweise dosisabhängig. Um die Wirksamkeit und Toxizität des Arzneimittels zu untersuchen, werden seine Wirkungen in einer Population getestet und gegen die Konzentration oder Dosis aufgetragen. Man erhält Sigmoidkurven (wenn die logarithmischen Konzentrationen auf der X-Achse aufgetragen werden), die die beobachteten Wirkungen widerspiegeln können.
In der folgenden Abbildung zeigt ein Sedativum/Hypnotikum beispielsweise die sedierende Wirkung in einem bestimmten Konzentrationsbereich und den Tod jenseits dieses Bereichs. Die Dosis (oder die Konzentration), bei der das Mittel bei 50 % der Bevölkerung die gewünschte Wirkung (Sedierung) zeigt, ist ED50.
Die Dosis (oder die Konzentration), bei der das Mittel bei 50 % der Bevölkerung die toxischen Wirkungen (z. B. Atemdepression) zeigt, ist TD50. Die Dosis (oder die Konzentration), bei der die Droge bei 50 % der Bevölkerung eine tödliche Wirkung (Tod) zeigt, ist LD50. (Sie wird für Tierversuche verwendet.)
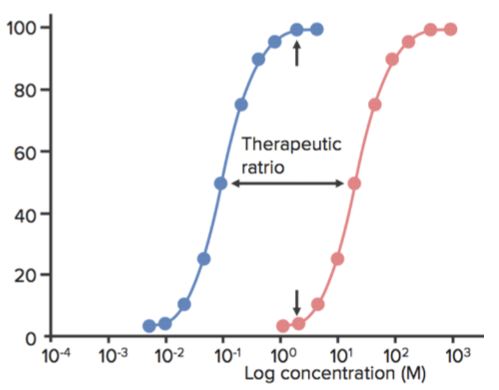
Das therapeutische Fenster ist der Dosisbereich zwischen der minimal wirksamen Konzentration und der minimal toxischen Konzentration. Der therapeutische Index (TI) ist definiert als das Verhältnis zwischen TD50 und ED50 oder TI=TD50/ED50.
- Dies wird manchmal auch als therapeutisches Verhältnis bezeichnet.
- Wenn ein Arzneimittel einen niedrigen TI-Wert hat, bedeutet dies, dass eine Erhöhung der Dosis leicht toxische (oder tödliche) Wirkungen hervorrufen kann.
- Trizyklische Antidepressiva und Lithium haben beispielsweise einen niedrigen TI-Wert, weshalb ihre Dosis bei Patienten sehr sorgfältig überwacht werden muss. Imipramin zum Beispiel kann tödlich sein, wenn es in der 5-6-fachen Tageshöchstdosis verabreicht wird.
- Je höher der TI, desto sicherer ist das Medikament (Beispiel: Penicillin).
Mit Lecturio für das Medizinstudium und die Prüfungen lernen.
- USMLE Step 1
- USMLE Step 2
- COMLEX Level 1
- COMLEX Level 2
- ENARM
- NEET