Imagen : «334 » por Evan Blaser. Licencia: CC BY-SA 2.0
- Definiciones importantes para entender la farmacodinámica
- Receptores
- Efectores
- Afinidad
- Selectividad
- Especificidad
- Agonismo
- Antagonismo
- Sinergismo
- Eficacia
- Potencia
- Excreción
- Eliminación
- Receptores y efectores
- Receptores
- Efectores
- Preglamentación y desreglamentación
- Preglamentación
- Desregulación
- Curvas dosis-respuesta
- Curvas de unión
- Receptores de reserva
- Toxicidad, curvas de toxicidad y relación terapéutica
Definiciones importantes para entender la farmacodinámica
Receptores
Un receptor es aquel componente (macromolécula) de una célula (sobre o dentro de la célula) que interactúa con el fármaco, y esta interacción conduce a una cadena de acontecimientos que alteran la actividad de la célula.
Efectores
Los efectores son moléculas que actúan en respuesta al fármaco (o más precisamente, al complejo fármaco-receptor) y participan en la mencionada cadena de acontecimientos intracelulares que conducen a los efectos del fármaco.
Afinidad
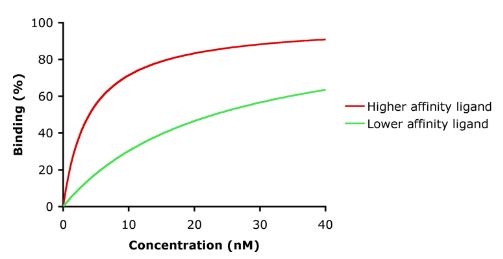
La afinidad es la medida de la fuerza de la unión entre el fármaco y su receptor. La afinidad de un fármaco con su receptor ayuda a determinar la dosis del fármaco: una afinidad baja indicaría la necesidad de una dosis más alta para formar suficientes complejos fármaco-receptor que den lugar a un efecto significativo (véase la figura).
Selectividad
La selectividad es la preferencia del fármaco por un receptor o su subtipo (en comparación con otros receptores u otros subtipos). En otras palabras, es la capacidad del fármaco para producir un efecto farmacológico sobre otros. Si un fármaco es selectivo, se unirá preferentemente a un receptor, pero puede unirse a otros aumentando su concentración. Por ejemplo, el verapamilo normalmente bloquea los canales Ca, pero puede bloquear los canales Na a altas concentraciones.
Especificidad
La especificidad es la capacidad del fármaco de unirse a un solo receptor. La atropina es técnicamente específica porque actúa sólo en los receptores de acetilcolina (ACh). Sin embargo, la atropina no es selectiva porque se une a todos los subtipos y provoca innumerables efectos farmacológicos.
Agonismo
Cuando el fármaco interactúa con el receptor para producir una serie de acontecimientos que conducen a un efecto farmacológico, se denomina agonismo. Por ejemplo, la isoprenalina es un agonista del adrenorreceptor β.
Antagonismo
Cuando el fármaco interactúa con el receptor pero no produce ninguna serie de acontecimientos, con lo que «bloquea» la acción potencial (física o químicamente), se denomina antagonismo. Por ejemplo, el propranolol es un antagonista del adrenorreceptor β.
Los antagonistas pueden ser competitivos (al unirse de forma reversible al receptor) o no competitivos (se unen de forma irreversible al receptor o cambiando alostéricamente la conformación de la proteína/enzima). La acción de un inhibidor competitivo puede revertirse aumentando la concentración del agonista, mientras que esto no ocurre con los inhibidores no competitivos.
Sinergismo
Cuando la acción de un fármaco se incrementa cuando se administra en presencia de otro fármaco, se denomina sinergismo. Los ejemplos son la aspirina y el paracetamol.
Eficacia
La eficacia es la propiedad del fármaco de producir el efecto biológico (deseado). La eficacia se utiliza a menudo cuando se comparan o evalúan fármacos en ensayos clínicos.
Potencia
La potencia es la concentración de un fármaco necesaria para producir un efecto de una intensidad determinada.
- Suele calcularse como la concentración (o dosis) necesaria para producir el 50% del efecto máximo del fármaco (EC>50).
- EC50 se utiliza sólo en estudios in vitro. Cuando la potencia de un fármaco se mide en una población (estudios en animales o poblaciones humanas), se utiliza otro parámetro denominado dosis efectiva media o ED50. La ED50 es la dosis que produce el efecto deseado (típicamente, un efecto cuántico) en el 50% de la población.
Al aplicar estos conceptos al ámbito clínico, la potencia simplemente indica la dosis del fármaco, mientras que la eficacia indica la magnitud de la respuesta (independientemente de la dosis).
Excreción
La excreción es la eliminación irreversible del fármaco del organismo. La excreción se produce principalmente desde el hígado y los riñones, pero también pueden intervenir otros órganos (por ejemplo, los pulmones).
Eliminación
La eliminación es el proceso de inactivación del fármaco con/sin eliminación real del organismo. La eliminación puede producirse por excreción o por metabolismo/biotransformación del fármaco. Así, mientras que la eliminación es la mera inactivación del fármaco, la excreción es la transferencia física del fármaco (en forma activa o inactiva) desde la circulación a los fluidos de excreción, como la orina y la bilis.
Receptores y efectores
Receptores
Existen cinco tipos básicos de receptores transmembrana, que actúan, tras unirse al fármaco o al ligando, de diferentes maneras (ver figuras).
![]()
- Receptores intracelulares: Son receptores proteicos que requieren que el fármaco atraviese la membrana plasmática; por tanto, el fármaco debe ser lipofílico. Los esteroides, por ejemplo, actúan por este mecanismo.
- Enzimas transmembrana: Un fármaco se une al componente extracelular de este receptor, que activa una reacción enzimática en el componente intracelular.
- Tirosina quinasa: Cuando un fármaco se une al componente extracelular de este receptor, conduce a la dimerización de las dos partes del receptor intracelularmente. Esta dimerización activa las enzimas tirosina quinasa, lo que conduce a la fosforilación de las moléculas de tirosina en las proteínas diana. Las hormonas del crecimiento y los interferones actúan a través de los receptores JAK-STAT-quinasa.
- Canales iónicos activados por ligando: Estos canales iónicos están cerrados por el ligando, es decir, están cerrados hasta que el receptor se une al fármaco, que entonces permite el paso de iones específicos. Por ejemplo, los fármacos que estimulan los receptores GABA en las neuronas provocan la afluencia de cloruro (lo que lleva a la hiperpolarización y, por tanto, a la inhibición).
- Receptores acoplados a proteínas G: De forma similar a los receptores de tirosina quinasa, la unión del fármaco al receptor conduce a la interacción de la proteína G con el receptor. Esta proteína G activada conduce entonces a la respuesta farmacológica deseada a través de una o una serie de moléculas efectoras o segundos mensajeros. Los receptores acoplados a la proteína G son tipos comunes de receptores en el organismo.
Efectores
Transducción de señales
Un fármaco, cuando se une al receptor (extracelularmente), actúa como «señal» para el evento o eventos subsiguientes (intracelularmente) que finalmente conducen a la respuesta farmacológica. Esta transmisión de la señal se denomina transducción de la señal, y la cadena intracelular de acontecimientos que intervienen en este proceso se denomina cascadas de transducción de la señal.
En esa cadena de acontecimientos pueden intervenir varias moléculas (denominadas segundos mensajeros). La función de las cascadas es amplificar la señal del fármaco. Como se ha mencionado anteriormente, los receptores acoplados a la proteína G actúan a través de segundos mensajeros. Una proteína G contiene una subunidad α que une el trifosfato de guanosina (GTP) y las subunidades β y γ que anclan la proteína en la membrana.
- La subunidad α se disocia con las otras dos tras la unión del fármaco al receptor, y los dos complejos disociados pueden unirse a otras enzimas para generar las cascadas.
- Un mecanismo común es la unión de la subunidad α a la enzima adenilil ciclasa y su activación, lo que hace que el ATP en la célula se convierta en, es AMP cíclico (cAMP). (La activación es causada por el tipo Gs de la proteína G; Gi inhibe la adenilil ciclasa). El aumento de las concentraciones de fosfato inorgánico puede unirse a la diana o a otras moléculas efectoras intermedias mediante la fosforilación de las proteínas.
Otros ejemplos de segundos mensajeros son el diacilglicerol (DAG), el inositol 1,4,5-trifosfato (IP3), ambos activados por la proteína G de tipo Gq; debido a que los receptores de la proteína G utilizan la fosforilación como mecanismo de acción, suelen ser susceptibles al fenómeno de desensibilización.
Preglamentación y desreglamentación
Preglamentación
Preglamentación (es decir, aumento del número) de receptores se produce cuando la actividad del receptor es inferior a la habitual (por ejemplo, debido a la administración a largo plazo de un antagonista). Por ejemplo, la administración de betabloqueantes regula al alza los adrenorreceptores β. Así, si los β-bloqueantes se suspenden bruscamente, puede causar hipertensión de rebote debido a la estimulación repentina de un gran número de β adrenorreceptores.
Desregulación
La desregulación (es decir, la disminución del número) es la inversa de la regulación al alza. Se produce debido a la administración repetida o a largo plazo de un agonista. Junto con la regulación a la baja, también puede producirse la desensibilización del receptor al fármaco. Se trata de una alteración fisicoquímica en el receptor que hace que no responda al fármaco; también se denomina taquifilaxia y se observa, por ejemplo, en el uso crónico de fármacos.
Curvas dosis-respuesta
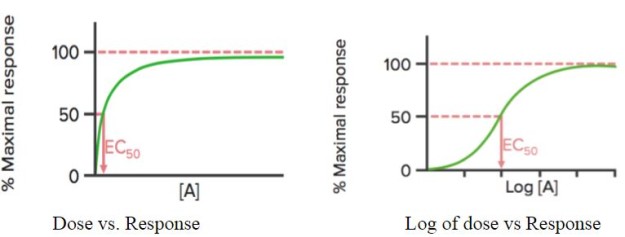
Cuando se traza la respuesta del fármaco frente a su dosis , muestra una curva hiperbólica; si se utiliza el logaritmo de la dosis (log), se observa una curva sigmoidal.
Esta curva puede utilizarse para detectar la concentración efectiva (o dosis) del fármaco a la que se obtiene el 50% de la respuesta máxima (EC50) del mismo. Como se ha mencionado anteriormente, la EC50 se utiliza para calcular/comparar la potencia de los fármacos.
Emax es la concentración (mínima) a la que se observa el máximo efecto del fármaco. En Emax, todos los receptores están ocupados por el fármaco.
Curvas de unión
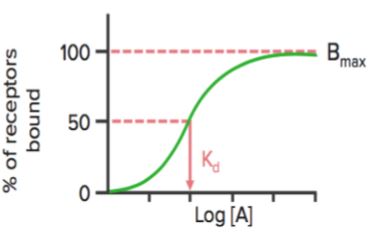
Cuando se representa el estado de unión de los receptores (% de receptores unidos) frente a la concentración del fármaco ( o logaritmo), muestra una curva hiperbólica (o sigmoidal) similar.
Usando este gráfico, podemos calcular la concentración efectiva a la que se unen el 50% de los receptores (Kd). Kd muestra la afinidad de unión del fármaco: un valor alto de Kd indica que se necesita una concentración elevada del fármaco para unir el 50% de los receptores, lo que indica una baja afinidad.
Bmax es la unión máxima del fármaco. Es la concentración, expresada en picomoles por mg de proteína, a la que todos los receptores específicos se unen al fármaco. Como resultado, Bmáx puede utilizarse para calcular la densidad del sitio del receptor en una preparación particular.
Receptores de reserva
Cuando un agonista se une a un receptor, puede producir una serie de eventos que finalmente conducen a la respuesta farmacológica. Esta serie de acontecimientos puede amplificar el efecto del fármaco de manera que sólo se necesite una cantidad relativamente pequeña de los complejos fármaco-receptor para dar lugar a una respuesta máxima. En este caso, cuando se observa la respuesta máxima, un porcentaje de receptores permanece desocupado por el fármaco. Estos receptores se denominan receptores de reserva.
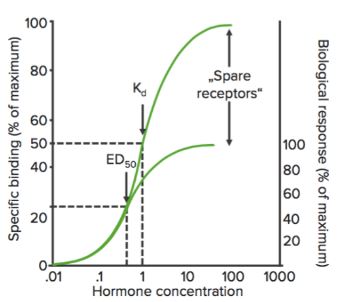
Los receptores de reserva, al menos en teoría, aumentan la sensibilidad del fármaco.
Toxicidad, curvas de toxicidad y relación terapéutica
Los efectos adversos de un fármaco suelen depender de la dosis. Para investigar la eficacia y la toxicidad del fármaco, se prueban sus efectos en una población y se trazan en función de la concentración o la dosis. Se obtienen curvas sigmoides (si se toman las concentraciones logarítmicas en el eje X), que pueden reflejar los efectos observados.
Por ejemplo, en la siguiente figura, un agente sedante/hipnótico muestra la acción sedante en un determinado rango de concentración y la muerte más allá de ese rango. La dosis (o la concentración) a la que el fármaco muestra el efecto deseado (sedación) en el 50% de la población es ED50.
La dosis (o la concentración) a la que el fármaco muestra los efectos tóxicos (por ejemplo, depresión respiratoria) en el 50% de la población es TD50. La dosis (o la concentración) a la que el fármaco muestra el efecto letal (muerte) en el 50% de la población es la DL50. (Se utiliza para los experimentos con animales.)
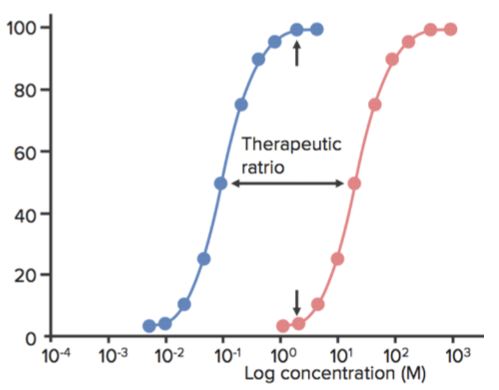
La ventana terapéutica es el rango de dosis entre la concentración mínima efectiva y la concentración mínima tóxica. El índice terapéutico (IT) se define como la relación entre la TD50 y la ED50 o IT=TD50/ED50.
- A veces también se denomina relación terapéutica.
- Cuando un fármaco tiene un IT bajo, significa que el aumento de su dosis puede producir fácilmente efectos tóxicos (o letales).
- Los antidepresivos tricíclicos y el litio, por ejemplo, tienen un IT bajo, y su dosis debe, por tanto, controlarse muy cuidadosamente en los pacientes. La imipramina, por ejemplo, puede ser mortal si se administra entre 5 y 6 veces la dosis máxima diaria.
- Cuanto mayor sea el IT, más seguro será el fármaco (el ejemplo es la penicilina).
Estudia para la facultad de medicina y los exámenes con Lecturio.
- USMLE Paso 1
- USMLE Paso 2
- COMLEX Nivel 1
- COMLEX Nivel 2
- ENARM
- NEET
.