Stabilité des intermédiaires de carbocation
Nous savons que l’étape limitant la vitesse d’une réaction SN1 est la première étape – la formation de cet intermédiaire de carbocation. La vitesse de cette étape – et donc, la vitesse de la réaction de substitution globale – dépend de l’énergie d’activation pour le processus dans lequel la liaison entre le carbone et le groupe partant se rompt et un carbocation se forme. Selon le postulat de Hammond (section 6.2B), plus l’intermédiaire carbocation est stable, plus cette première étape de rupture de la liaison se produira rapidement. En d’autres termes, la probabilité qu’une réaction de substitution nucléophile se déroule par un mécanisme dissociatif (SN1) dépend dans une large mesure de la stabilité de l’intermédiaire carbocationique qui se forme.
La question critique devient maintenant : qu’est-ce qui stabilise un carbocation ?
Si donc il faut un groupe attracteur d’électrons pour stabiliser une charge négative, qu’est-ce qui stabilisera une charge positive ? Un groupe donneur d’électrons !
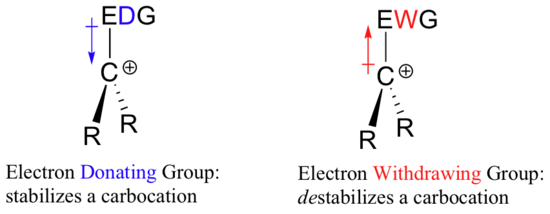
Une espèce chargée positivement telle qu’un carbocation est très pauvre en électrons, et donc tout ce qui donne de la densité électronique au centre de la pauvreté électronique aidera à la stabiliser. Inversement, un carbocation sera déstabilisé par un groupe attracteur d’électrons.
Les groupes alkyles – méthyle, éthyle et autres – sont des groupes donneurs d’électrons faibles, et stabilisent donc les carbocations proches. Ce que cela signifie, c’est qu’en général, les carbocations plus substituées sont plus stables : une carbocation tert-butyle, par exemple, est plus stable qu’une carbocation isopropyle. Les carbocations primaires sont très instables et ne sont pas souvent observées comme intermédiaires de réaction ; les carbocations méthyliques sont encore moins stables.
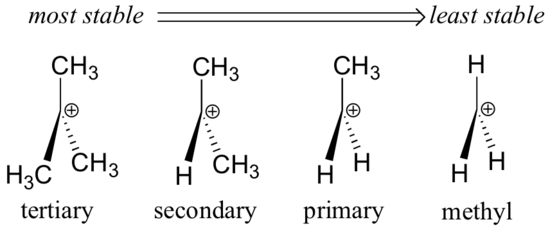
Les groupes alkyles sont donneurs d’électrons et stabilisent les carbocations car les électrons autour des carbones voisins sont attirés vers la charge positive proche, réduisant ainsi légèrement la pauvreté en électrons du carbone chargé positivement.
Il n’est cependant pas exact de dire que les carbocations à plus forte substitution sont toujours plus stables que celles à moindre substitution. Tout comme les groupes donneurs d’électrons peuvent stabiliser un carbocation, les groupes arracheurs d’électrons agissent pour déstabiliser les carbocations. Les groupes carbonyles attirent les électrons par effet inductif, en raison de la polarité de la double liaison C=O. Il est possible de démontrer en laboratoire (voir section 16.1D) que le carbocation A ci-dessous est plus stable que le carbocation B, même si A est un carbocation primaire et B un carbocation secondaire.
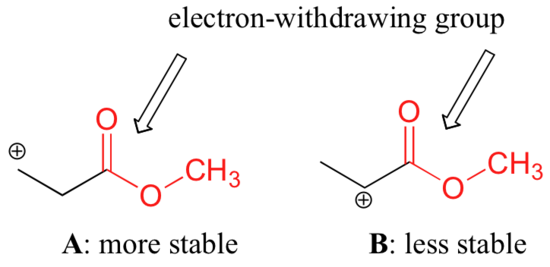
La différence de stabilité peut être expliquée en considérant l’effet inductif attracteur d’électrons du carbonyle de l’ester. Rappelons que les effets inductifs – qu’ils soient donneurs ou arracheurs d’électrons – sont relayés par des liaisons covalentes et que la force de l’effet diminue rapidement lorsque le nombre de liaisons intermédiaires augmente. En d’autres termes, l’effet diminue avec la distance. Dans l’espèce B, la charge positive est plus proche du groupe carbonyle, donc l’effet déstabilisateur de retrait d’électrons est plus fort que dans l’espèce A.
Dans le prochain chapitre, nous verrons comment l’effet déstabilisateur de carbocation des substituants fluorés de retrait d’électrons peut être utilisé dans des expériences conçues pour répondre à la question de savoir si une réaction de substitution nucléophile biochimique est SN1 ou SN2.
La stabilisation d’un carbocation peut également se produire par des effets de résonance, et comme nous l’avons déjà discuté dans le chapitre acide-base, les effets de résonance en règle générale sont plus puissants que les effets inductifs. Considérons le cas simple d’un carbocation benzylique :
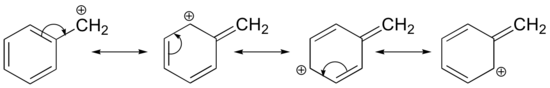
Ce carbocation est comparativement stable. Dans ce cas, le don d’électrons est un effet de résonance. Trois structures de résonance supplémentaires peuvent être dessinées pour ce carbocation dans lequel la charge positive est située sur l’un des trois carbones aromatiques. La charge positive n’est pas isolée sur le carbone benzylique, elle est plutôt délocalisée autour de la structure aromatique : cette délocalisation de la charge entraîne une stabilisation importante. Par conséquent, les carbocations benzyliques et allyliques (où le carbone chargé positivement est conjugué à une ou plusieurs doubles liaisons non aromatiques) sont nettement plus stables que les carbocations alkyle même tertiaires.
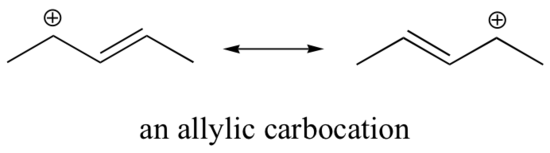
Parce que les hétéroatomes tels que l’oxygène et l’azote sont plus électronégatifs que le carbone, on pourrait s’attendre à ce qu’ils soient par définition des groupes attracteurs d’électrons qui déstabilisent les carbocations. En fait, le contraire est souvent vrai : si l’atome d’oxygène ou d’azote est dans la bonne position, l’effet global est la stabilisation des carbocations. Cela est dû au fait que, bien que ces hétéroatomes soient des groupes attracteurs d’électrons par induction, ils sont des groupes donneurs d’électrons par résonance, et c’est cet effet de résonance qui est le plus puissant. (Nous avons déjà rencontré cette même idée en examinant l’acidité et la basicité relatives des phénols et des amines aromatiques dans la section 7.4). Considérons les deux paires d’espèces de carbocation ci-dessous :
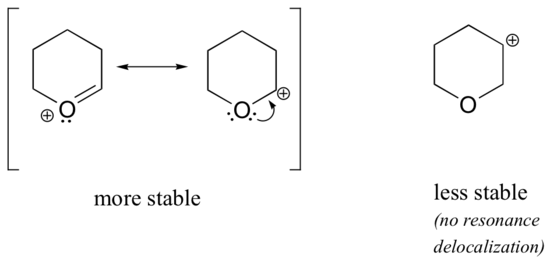
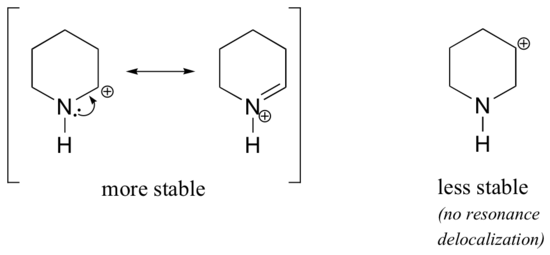
Dans les carbocations les plus stables, l’hétéroatome agit comme un groupe donneur d’électrons par résonance : en effet, le doublet solitaire sur l’hétéroatome est disponible pour délocaliser la charge positive. Dans les carbocations moins stables, le carbone chargé positivement est à plus d’une liaison de l’hétéroatome, et donc aucun effet de résonance n’est possible. En fait, dans ces espèces de carbocations, les hétéroatomes déstabilisent réellement la charge positive, car ils retirent des électrons par induction.
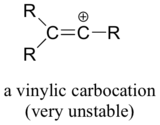
Enfin, les carbocations vinyliques, dans lesquelles la charge positive réside sur un carbone à double liaison, sont très instables et donc peu susceptibles de se former comme intermédiaires dans une quelconque réaction.
Exemple 7.9.1
Dans laquelle des structures ci-dessous la carbocation devrait-elle être plus stable ? Expliquez.
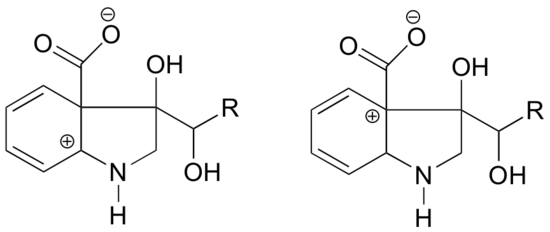
Réponse
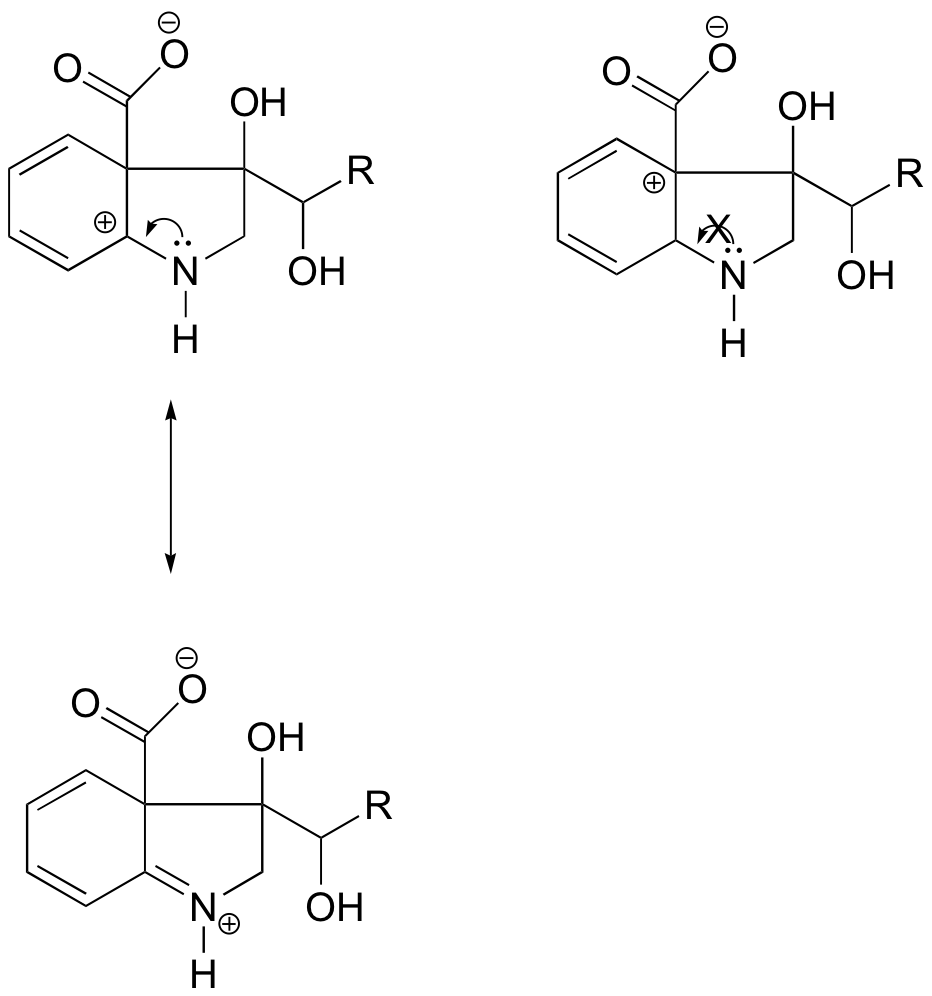
Dans le carbocation de gauche, la charge positive est située dans une position par rapport à l’azote telle que la paire d’électrons solitaires sur l’azote peut être donnée pour remplir l’orbitale vide. Ceci n’est pas possible pour l’espèce de carbocation à droite.
Pour la plupart, les carbocations sont des espèces intermédiaires transitoires à très haute énergie dans les réactions organiques. Cependant, il existe quelques exemples inhabituels de carbocations très stables qui prennent la forme de sels organiques. Le violet cristal est le nom commun du sel de chlorure du carbocation dont la structure est illustrée ci-dessous. Remarquez les possibilités structurelles de délocalisation importante de la charge positive par résonance, et la présence de trois groupes amine donneurs d’électrons.
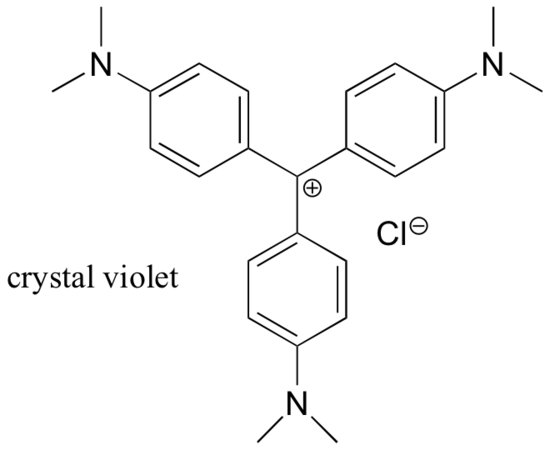
Exemple 7.9.2
Dessinez une structure de résonance du cation cristal violet dans laquelle la charge positive est délocalisée vers l’un des atomes d’azote.
Réponse 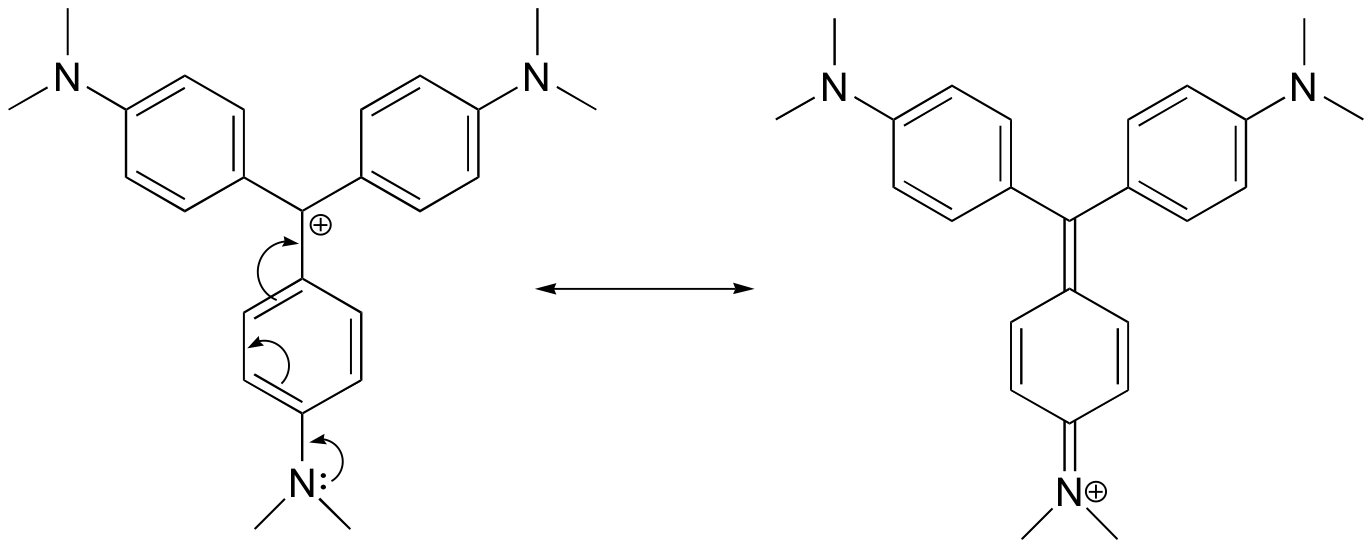
Lorsqu’on envisage la possibilité qu’une réaction de substitution nucléophile se déroule par une voie SN1, il est essentiel d’évaluer la stabilité de l’intermédiaire carbocation hypothétique. Si cet intermédiaire n’est pas suffisamment stable, un mécanisme SN1 doit être considéré comme peu probable, et la réaction se déroule probablement par un mécanisme SN2. Dans le prochain chapitre, nous verrons plusieurs exemples de réactions SN1 biologiquement importantes dans lesquelles l’intermédiaire chargé positivement est stabilisé par des effets d’induction et de résonance inhérents à sa propre structure moléculaire.
Exemple 7.9.3
Déterminez quel carbocation dans chaque paire ci-dessous est le plus stable, ou si l’on s’attend à ce qu’ils soient approximativement égaux. Expliquez votre raisonnement.
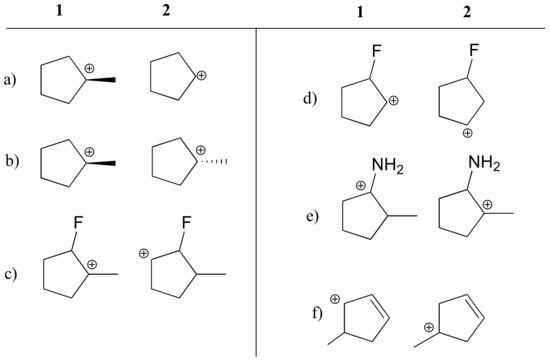
Réponse
a) 1 (carbocation tertiaire vs secondaire)
b) égal
c) 1 (carbocation tertiaire vs. secondaire)
d) 2 (la charge positive est plus loin du fluor qui attire les électrons)
e) 1 (le couple isolé sur l’azote peut donner des électrons par résonance)
f) 1 (carbocation allylique – la charge positive peut être délocalisée vers un deuxième carbone)
.