Image : « 334 » par Evan Blaser. Licence : CC BY-SA 2.0
- Définitions importantes pour comprendre la pharmacodynamique
- Récepteurs
- Effecteurs
- Affinité
- Sélectivité
- Spécificité
- Agonisme
- Antagonisme
- Synergie
- Efficacité
- Puissance
- Excrétion
- Elimination
- Récepteurs et effecteurs
- Récepteurs
- Effecteurs
- Upregulation et downregulation
- Upregulation
- Downregulation
- Courbes dose-réponse
- Courbes de liaison
- Sparez les récepteurs
- Toxicité, courbes de toxicité et rapport thérapeutique
Définitions importantes pour comprendre la pharmacodynamique
Récepteurs
Un récepteur est le composant (macromolécule) d’une cellule (sur ou à l’intérieur de la cellule) qui interagit avec le médicament, et cette interaction entraîne une chaîne d’événements qui modifient l’activité de la cellule.
Effecteurs
Les effecteurs sont des molécules qui agissent en réponse au médicament (ou plus précisément au complexe médicament-récepteur) et participent à la chaîne précitée d’événements intracellulaires conduisant aux effets du médicament.
Affinité
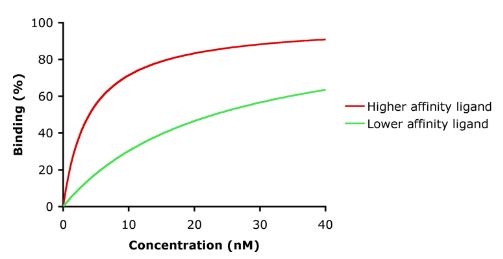
L’affinité est la mesure de la force du lien entre le médicament et son récepteur. L’affinité d’un médicament pour son récepteur aide à déterminer la dose du médicament : une faible affinité indiquerait la nécessité d’une dose plus élevée pour former suffisamment de complexes médicament-récepteur qui conduiraient à un effet significatif (voir figure).
Sélectivité
La sélectivité est la préférence du médicament pour un récepteur ou son sous-type (par rapport à d’autres récepteurs ou d’autres sous-types). En d’autres termes, il s’agit de la capacité du médicament à produire un effet pharmacologique plutôt que d’autres. Si un médicament est sélectif, il se fixera de préférence à un récepteur, mais il peut se fixer à d’autres en augmentant sa concentration. Par exemple, le vérapamil bloque normalement les canaux Ca, mais il peut bloquer les canaux Na à des concentrations élevées.
Spécificité
La spécificité est la capacité du médicament à se lier à un seul récepteur. L’atropine est techniquement spécifique car elle agit uniquement sur les récepteurs de l’acétylcholine (ACh). Cependant, l’atropine n’est pas sélective car elle se lie à tous les sous-types et provoque une myriade d’effets pharmacologiques.
Agonisme
Lorsque le médicament interagit avec le récepteur pour produire une série d’événements conduisant à un effet pharmacologique, on parle d’agonisme. Par exemple, l’isoprénaline est un agoniste des β-adrénorécepteurs.
Antagonisme
Lorsque le médicament interagit avec le récepteur mais ne produit aucune série d’événements, » bloquant » ainsi l’action potentielle (physiquement ou chimiquement), on parle d’antagonisme. Par exemple, le propranolol est un antagoniste des β-adrénorécepteurs.
Les antagonistes peuvent être compétitifs (en se liant de manière réversible au récepteur) ou non compétitifs (en se liant de manière irréversible au récepteur ou en modifiant de manière allostérique la conformation de la protéine/enzyme). L’action d’un inhibiteur compétitif peut être inversée en augmentant la concentration de l’agoniste, alors que ce n’est pas le cas pour les inhibiteurs non compétitifs.
Synergie
Lorsque l’action d’un médicament est augmentée lorsqu’il est administré en présence d’un autre médicament, on parle de synergie. Des exemples sont l’aspirine et le paracétamol.
Efficacité
L’efficacité est la propriété du médicament à produire l’effet biologique (souhaité). L’efficacité est souvent utilisée lors de la comparaison ou de l’évaluation des médicaments dans les essais cliniques.
Puissance
La puissance est la concentration d’un médicament nécessaire pour produire un effet d’une intensité spécifiée.
- Elle est généralement calculée comme la concentration (ou la dose) nécessaire pour produire 50% de l’effet maximal du médicament (CE>50).
- La CE50 est utilisée dans les études in-vitro uniquement. Lorsque la puissance d’un médicament est mesurée dans une population (études animales ou populations humaines), un autre paramètre appelé dose efficace médiane ou ED50 est utilisé. La DE50 est la dose qui produit l’effet désiré (typiquement, un effet quantique) chez 50 % de la population.
Lorsque l’on applique ces concepts au contexte clinique, la puissance indique simplement la dose du médicament, tandis que l’efficacité indique l’ampleur de la réponse (indépendamment de la dose).
Excrétion
L’excrétion est l’élimination irréversible du médicament de l’organisme. L’excrétion se produit principalement à partir du foie et des reins, mais d’autres organes (par exemple, les poumons) peuvent également être impliqués.
Elimination
L’élimination est le processus d’inactivation du médicament avec/sans élimination réelle du corps. L’élimination peut se produire en raison de l’excrétion ou du métabolisme/biotransformation du médicament. Ainsi, alors que l’élimination est une simple inactivation du médicament, l’excrétion est le transfert physique du médicament (forme active ou inactive du) de la circulation vers les liquides d’excrétion, tels que l’urine et la bile.
Récepteurs et effecteurs
Récepteurs
Il existe cinq types fondamentaux de récepteurs transmembranaires, qui agissent, après s’être liés au médicament ou au ligand, de différentes manières (voir figures).
![]()
- Récepteurs intracellulaires : Ce sont des récepteurs protéiques qui nécessitent que le médicament traverse la membrane plasmique ; le médicament doit donc être lipophile. Les stéroïdes, par exemple, agissent par ce mécanisme.
- Enzymes transmembranaires : Un médicament se lie au composant extracellulaire de ce récepteur, ce qui active une réaction enzymatique dans le composant intracellulaire.
- Tyrosine kinase : Lorsqu’un médicament se lie à la composante extracellulaire de ce récepteur, il entraîne une dimérisation des deux parties du récepteur au niveau intracellulaire. Cette dimérisation active les enzymes tyrosine kinase, entraînant ainsi la phosphorylation des molécules de tyrosine sur les protéines cibles. Les hormones de croissance et les interférons agissent par l’intermédiaire des récepteurs JAK-STAT-kinase.
- Canaux ioniques dépendant d’un ligand : Ces canaux ioniques sont ligand-gated, c’est-à-dire qu’ils sont fermés jusqu’à ce que le récepteur se lie au médicament, qui laisse alors passer des ions spécifiques. Par exemple, les médicaments qui stimulent les récepteurs GABA sur les neurones provoquent un afflux de chlorure (entraînant une hyperpolarisation et donc une inhibition).
- Récepteurs couplés aux protéines G : Comme pour les récepteurs à tyrosine kinase, la liaison médicament-récepteur entraîne l’interaction de la protéine G avec le récepteur. Cette protéine G activée entraîne ensuite la réponse pharmacologique souhaitée par le biais d’une ou plusieurs molécules effectrices ou seconds messagers. Les récepteurs couplés aux protéines G sont des types de récepteurs courants dans l’organisme.
Effecteurs
Transduction du signal
Un médicament, lorsqu’il se lie au récepteur (extracellulairement), agit comme un « signal » pour le ou les événements ultérieurs (intracellulairement) qui conduisent finalement à la réponse pharmacologique. Cette transmission du signal est appelée transduction du signal, et la chaîne intracellulaire des événements qui sont impliqués dans ce processus est appelée cascades de transduction du signal.
Un certain nombre de molécules (appelées seconds messagers) peuvent être impliquées dans ces chaînes d’événements. La fonction de ces cascades est d’amplifier le signal du médicament. Comme mentionné précédemment, les récepteurs couplés aux protéines G agissent via des seconds messagers. Une protéine G contient une sous-unité α qui se lie au guanosine triphosphate (GTP) et les sous-unités β et γ qui ancrent la protéine dans la membrane.
- La sous-unité α se dissocie avec les deux autres après la liaison médicament-récepteur, et les deux complexes dissociés peuvent se lier à d’autres enzymes pour générer les cascades.
- Un mécanisme commun est la liaison de la sous-unité α à l’enzyme adénylyl cyclase et son activation, ce qui entraîne la conversion de l’ATP dans la cellule en, est AMP cyclique (cAMP). (L’activation est provoquée par la protéine Gs de type G ; Gi inhibe l’adénylyl cyclase). L’augmentation des concentrations de phosphate inorganique peut se lier à la cible ou à d’autres molécules effectrices intermédiaires par phosphorylation des protéines.
D’autres exemples de seconds messagers sont le diacylglycérol (DAG), l’inositol 1,4,5-triphosphate (IP3), qui sont tous deux activés par la protéine G de type Gq ; comme les récepteurs de la protéine G utilisent la phosphorylation comme mécanisme d’action, ils sont souvent sensibles au phénomène de désensibilisation.
Upregulation et downregulation
Upregulation
Upregulation (c’est-à-dire, augmentation du nombre) de récepteurs se produit lorsque l’activité du récepteur est plus faible que d’habitude (par exemple, en raison de l’administration à long terme d’un antagoniste). Par exemple, l’administration de bêta-bloquants régule à la hausse les β-adrénorécepteurs. Ainsi, si les β-bloquants sont brusquement arrêtés, cela peut provoquer une hypertension de rebond en raison de la stimulation soudaine d’un grand nombre de β-adrénorécepteurs.
Downregulation
La downregulation (c’est-à-dire la diminution du nombre) est l’inverse de la upregulation. Elle se produit en raison de l’administration répétée ou à long terme d’un agoniste. Parallèlement à la dérégulation, une désensibilisation du récepteur au médicament peut également se produire. Il s’agit d’une altération physico-chimique du récepteur qui le rend insensible au médicament ; ce phénomène est également appelé tachyphylaxie et s’observe par exemple dans la consommation chronique de médicaments.
Courbes dose-réponse
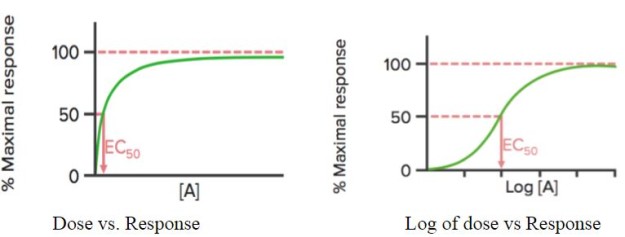
Lorsque la réponse du médicament est tracée en fonction de sa dose , elle montre une courbe hyperbolique ; si le logarithme de la dose (log) est utilisé, on observe une courbe sigmoïdale.
Cette courbe peut être utilisée pour détecter la concentration efficace (ou dose) du médicament à laquelle 50% de la réponse maximale (EC50) du médicament est obtenue. Comme mentionné précédemment, la CE50 est utilisée pour calculer/comparer la puissance des médicaments.
Emax est la concentration (minimale) à laquelle l’effet maximal du médicament est observé. A Emax, tous les récepteurs sont occupés par le médicament.
Courbes de liaison
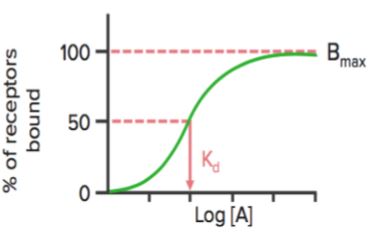
Lorsque l’état de liaison des récepteurs (% de récepteurs liés) est tracé en fonction de la concentration du médicament ( ou log), il montre une courbe hyperbolique (ou sigmoïdale) similaire.
En utilisant ce graphique, nous pouvons calculer la concentration efficace à laquelle 50% des récepteurs sont liés (Kd). Le Kd indique l’affinité de liaison du médicament – une valeur élevée du Kd indique qu’une concentration élevée du médicament est nécessaire pour lier 50% des récepteurs, indiquant ainsi une faible affinité.
Bmax est la liaison maximale du médicament. C’est la concentration, exprimée en picomoles par mg de protéine, à laquelle tous les récepteurs spécifiques sont liés au médicament. Par conséquent, Bmax peut être utilisé pour calculer la densité du site récepteur dans une préparation particulière.
Sparez les récepteurs
Lorsqu’un agoniste se lie à un récepteur, il peut produire une série d’événements qui conduisent finalement à la réponse pharmacologique. Cette série d’événements peut amplifier l’effet du médicament de telle sorte que seule une quantité relativement faible des complexes médicament-récepteur peut être nécessaire pour conduire à une réponse maximale. Dans ce cas, lorsque la réponse maximale est observée, un pourcentage de récepteurs reste inoccupé par le médicament. Ces récepteurs sont appelés récepteurs de réserve.
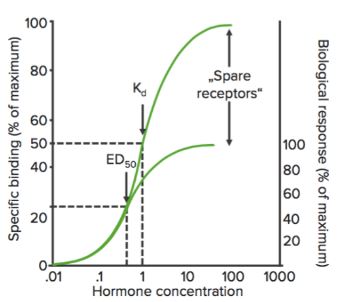
Les récepteurs de réserve, au moins théoriquement, augmentent la sensibilité du médicament.
Toxicité, courbes de toxicité et rapport thérapeutique
Les effets indésirables d’un médicament sont généralement dépendants de la dose. Pour étudier l’efficacité et la toxicité du médicament, ses effets sont testés dans une population et tracés en fonction de la concentration ou de la dose. On obtient des courbes sigmoïdes (si l’on prend les concentrations logarithmiques en abscisse) qui peuvent refléter les effets observés.
Par exemple, dans la figure suivante, un agent sédatif/hypnotique montre l’action sédative à une certaine gamme de concentration et la mort au-delà de cette gamme. La dose (ou la concentration) à laquelle le médicament présente l’effet désiré (sédation) chez 50% de la population est ED50.
La dose (ou la concentration) à laquelle le médicament présente les effets toxiques (par exemple, dépression respiratoire) chez 50% de la population est TD50. La dose (ou la concentration) à laquelle le médicament présente l’effet létal (mort) chez 50% de la population est la DL50. (Cette valeur est utilisée pour les expériences sur les animaux.)
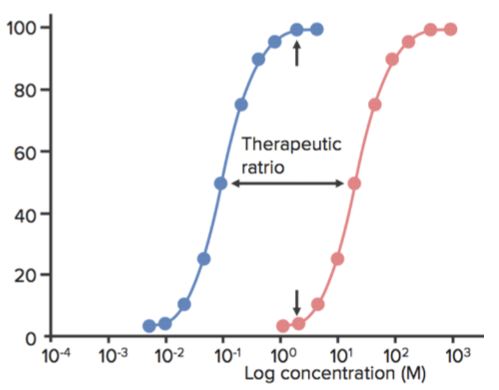
La fenêtre thérapeutique est l’intervalle de dose entre la concentration minimale efficace et la concentration minimale toxique. L’index thérapeutique (TI) est défini comme le rapport entre la TD50 et la ED50 ou TI=TD50/ED50.
- Ceci est parfois aussi appelé ratio thérapeutique.
- Quand un médicament a un TI faible, cela signifie que l’augmentation de sa dose peut facilement produire des effets toxiques (ou mortels).
- Les antidépresseurs tricycliques et le lithium, par exemple, ont un TI faible, et leur dose doit, par conséquent, être surveillée très attentivement chez les patients. L’imipramine, par exemple, peut être mortelle si elle est administrée à 5-6 fois la dose quotidienne maximale.
- Plus le TI est élevé, plus le médicament est sûr (l’exemple est la pénicilline).
Etudiez pour la faculté de médecine et les conseils avec Lecturio.
- USMLE Step 1
- USMLE Step 2
- COMLEX Niveau 1
- COMLEX Niveau 2
- ENARM
- NEET
.