Immagine : “334 ” di Evan Blaser. Licenza: CC BY-SA 2.0
- Definizioni importanti per capire la farmacodinamica
- Ricettori
- Effettori
- Affinità
- Selettività
- Specificità
- Agonismo
- Antagonismo
- Sinergismo
- Efficacia
- Potenza
- Escrezione
- Eliminazione
- Ricettori ed effettori
- Ricettori
- Effettori
- Upregolazione e Downregulation
- Upregolazione
- Downregulation
- Curve dose-risposta
- Curve di legame
- Recettori di riserva
- Tossicità, curve di tossicità e rapporto terapeutico
Definizioni importanti per capire la farmacodinamica
Ricettori
Un recettore è quel componente (macromolecola) di una cellula (sulla o dentro la cellula) che interagisce con il farmaco, e questa interazione porta ad una catena di eventi che alterano l’attività della cellula.
Effettori
Gli effettori sono molecole che agiscono in risposta al farmaco (o più precisamente, al complesso farmaco-recettore) e partecipano alla suddetta catena di eventi intracellulari che portano agli effetti del farmaco.
Affinità
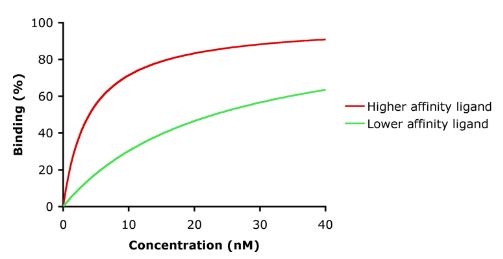
L’affinità è la misura della forza del legame tra il farmaco e il suo recettore. L’affinità di un farmaco al suo recettore aiuta a determinare la dose del farmaco: una bassa affinità indicherebbe la necessità di una dose più alta per formare abbastanza complessi farmaco-recettore che porterebbero a un effetto significativo (vedi figura).
Selettività
La selettività è la preferenza del farmaco per un recettore o un suo sottotipo (rispetto ad altri recettori o altri sottotipi). In altre parole, è la capacità del farmaco di produrre un effetto farmacologico rispetto ad altri. Se un farmaco è selettivo, si legherà preferibilmente a un recettore, ma può legarsi ad altri aumentandone la concentrazione. Per esempio, il verapamil blocca normalmente i canali Ca, ma può bloccare i canali Na ad alte concentrazioni.
Specificità
La specificità è la capacità del farmaco di legarsi a un solo recettore. L’atropina è tecnicamente specifica perché agisce solo ai recettori dell’acetilcolina (ACh). Tuttavia, l’atropina non è selettiva perché si lega a tutti i sottotipi e causa una miriade di effetti farmacologici.
Agonismo
Quando il farmaco interagisce con il recettore per produrre una serie di eventi che portano a un effetto farmacologico, si parla di agonismo. Per esempio, l’isoprenalina è un agonista del β adrenorecettore.
Antagonismo
Quando il farmaco interagisce con il recettore ma non produce alcuna serie di eventi, “bloccando” così la potenziale azione (fisicamente o chimicamente), si parla di antagonismo. Per esempio, il propranololo è un antagonista del β adrenorecettore.
Gli antagonisti possono essere competitivi (legandosi reversibilmente al recettore) o non competitivi (legandosi irreversibilmente al recettore o cambiando allostericamente la conformazione della proteina/enzima). L’azione di un inibitore competitivo può essere invertita aumentando la concentrazione dell’agonista, mentre questo non è il caso degli inibitori non competitivi.
Sinergismo
Quando l’azione di un farmaco è aumentata se somministrato in presenza di un altro farmaco, si parla di sinergia. Esempi sono l’aspirina e il paracetamolo.
Efficacia
L’efficacia è la proprietà del farmaco di produrre l’effetto biologico (desiderato). L’efficacia è spesso usata quando si confrontano o si valutano i farmaci negli studi clinici.
Potenza
La potenza è la concentrazione di un farmaco necessaria per produrre un effetto di un’intensità specificata.
- Di solito è calcolata come la concentrazione (o dose) necessaria per produrre il 50% dell’effetto massimo del farmaco (EC>50).
- EC50 è usata solo negli studi in vitro. Quando la potenza di un farmaco è misurata in una popolazione (studi su animali o popolazioni umane), si usa un altro parametro chiamato dose efficace mediana o ED50. ED50 è la dose che produce l’effetto desiderato (tipicamente, un effetto quantico) nel 50% della popolazione.
Quando si applicano questi concetti alle impostazioni cliniche, la potenza indica semplicemente la dose del farmaco, mentre l’efficacia indica la grandezza della risposta (indipendentemente dalla dose).
Escrezione
L’escrezione è la rimozione irreversibile del farmaco dal corpo. L’escrezione avviene principalmente dal fegato e dai reni, ma anche altri organi (per esempio, i polmoni) possono essere coinvolti.
Eliminazione
L’eliminazione è il processo di inattivazione del farmaco con/senza effettiva rimozione dal corpo. L’eliminazione può avvenire a causa dell’escrezione o del metabolismo/biotrasformazione del farmaco. Così, mentre l’eliminazione è la semplice inattivazione del farmaco, l’escrezione è il trasferimento fisico del farmaco (in forma attiva o inattiva) dalla circolazione ai fluidi di escrezione, come l’urina e la bile.
Ricettori ed effettori
Ricettori
Ci sono cinque tipi fondamentali di recettori transmembrana, che agiscono, dopo il legame al farmaco o al ligando, in modi diversi (vedi figure).
![]()
- Recettori intracellulari: Si tratta di recettori proteici che richiedono che il farmaco attraversi la membrana plasmatica; pertanto, il farmaco deve essere lipofilo. Gli steroidi, per esempio, agiscono con questo meccanismo.
- Enzimi transmembrana: Un farmaco si lega alla componente extracellulare di questo recettore, che attiva una reazione enzimatica nella componente intracellulare.
- Tirosina chinasi: Quando un farmaco si lega alla componente extracellulare di questo recettore, porta alla dimerizzazione delle due parti del recettore intracellulare. Questa dimerizzazione attiva gli enzimi tirosin-chinasici, portando così alla fosforilazione delle molecole di tirosina sulle proteine bersaglio. Gli ormoni della crescita e gli interferoni agiscono attraverso i recettori JAK-STAT-chinasi.
- Canali ionici ligando-gati: Questi canali ionici sono ligand-gated, cioè, sono chiusi fino a quando il recettore si lega al farmaco, che poi permette il passaggio di ioni specifici. Per esempio, i farmaci che stimolano i recettori GABA sui neuroni causano l’afflusso di cloruro (portando all’iperpolarizzazione e quindi all’inibizione).
- Recettori accoppiati alle proteine G: Simile ai recettori tirosin-chinasici, il legame farmaco-recettore porta all’interazione della proteina G con il recettore. Questa proteina G attivata porta poi alla risposta farmacologica desiderata attraverso una o una serie di molecole effettrici o secondi messaggeri. I recettori accoppiati alle proteine G sono tipi comuni di recettori nel corpo.
Effettori
Trasduzione del segnale
Un farmaco, quando si lega al recettore (extracellulare), agisce come “segnale” per l’evento o gli eventi successivi (intracellulari) che alla fine porta alla risposta farmacologica. Questa trasmissione del segnale è chiamata trasduzione del segnale, e la catena intracellulare di eventi che sono coinvolti in questo processo è chiamata cascata di trasduzione del segnale.
Un certo numero di molecole (chiamate secondi messaggeri) può essere coinvolto in questa catena di eventi. La funzione delle cascate è quella di amplificare il segnale del farmaco. Come menzionato prima, i recettori accoppiati alle proteine G agiscono tramite secondi messaggeri. Una proteina G contiene una subunità α che lega il trifosfato di guanosina (GTP) e le subunità β e γ che ancorano la proteina nella membrana.
- La subunità α si dissocia con le altre due dopo il legame farmaco-recettore, e i due complessi dissociati possono legarsi ad altri enzimi per generare le cascate.
- Un meccanismo comune è il legame della subunità α all’enzima adenilciclasi e la sua attivazione, che causa la conversione dell’ATP nella cellula in AMP ciclico (cAMP). (L’attivazione è causata dal tipo Gs della proteina G; Gi inibisce l’adenilciclasi). L’aumento delle concentrazioni di fosfato inorganico può legarsi al bersaglio o ad altre molecole effettrici intermedie attraverso la fosforilazione delle proteine.
Altri esempi di secondi messaggeri sono il diacilglicerolo (DAG), l’inositolo 1,4,5-trifosfato (IP3), entrambi attivati dalla proteina G di tipo Gq; poiché i recettori delle proteine G utilizzano la fosforilazione come meccanismo d’azione, sono spesso soggetti al fenomeno della desensibilizzazione.
Upregolazione e Downregulation
Upregolazione
Upregolazione (cioè, aumento del numero) di recettori si verifica quando l’attività del recettore è inferiore al solito (ad esempio, a causa della somministrazione a lungo termine di un antagonista). Per esempio, la somministrazione di beta-bloccanti upregulates β adrenoreceptors. Così, se i β-bloccanti sono interrotti bruscamente, può causare ipertensione di rimbalzo a causa della stimolazione improvvisa di un gran numero di β adrenorecettori.
Downregulation
Downregulation (cioè, diminuzione nel numero) è l’inverso di upregulation. Si verifica a causa della somministrazione ripetuta o a lungo termine di un agonista. Insieme alla downregulation, può verificarsi anche la desensibilizzazione del recettore al farmaco. Questa è un’alterazione fisico-chimica nel recettore che lo rende non responsivo al farmaco; questo è anche chiamato tachifilassi e si vede nell’uso cronico di droga, per esempio.
Curve dose-risposta
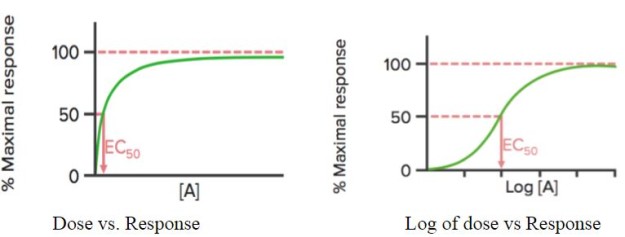
Quando la risposta del farmaco è tracciata contro la sua dose, mostra una curva iperbolica; se il log della dose (log) è usato, si vede una curva sigmoidale.
Questa curva può essere utilizzata per individuare la concentrazione efficace (o dose) del farmaco alla quale si ottiene il 50% della risposta massima (EC50) del farmaco. Come menzionato prima, l’EC50 è usato per calcolare/confrontare la potenza dei farmaci.
Emax è la concentrazione (minima) alla quale si osserva l’effetto massimo del farmaco. A Emax, tutti i recettori sono occupati dal farmaco.
Curve di legame
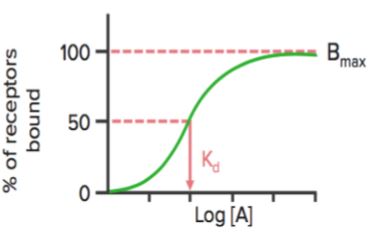
Quando lo stato di legame del recettore (% di recettori legati) è tracciato contro la concentrazione del farmaco ( o log), mostra una curva iperbolica (o sigmoidale) simile.
Utilizzando questo grafico, possiamo calcolare la concentrazione effettiva alla quale il 50% dei recettori sono legati (Kd). Kd mostra l’affinità di legame del farmaco – un alto valore di Kd indica che è necessaria un’alta concentrazione del farmaco per legare il 50% dei recettori, indicando così una bassa affinità.
Bmax è il legame massimo del farmaco. È la concentrazione, espressa in picomoli per mg di proteina, alla quale tutti i recettori specifici sono legati al farmaco. Di conseguenza, la Bmax può essere usata per calcolare la densità del sito recettoriale in una particolare preparazione.
Recettori di riserva
Quando un agonista si lega a un recettore, può produrre una serie di eventi che alla fine portano alla risposta farmacologica. Questa serie di eventi può amplificare l’effetto del farmaco in modo tale che solo una quantità relativamente piccola dei complessi farmaco-recettore può essere necessaria per portare ad una risposta massima. In questo caso, quando la risposta massima è vista, una percentuale di recettori rimane non occupata dal farmaco. Questi recettori sono chiamati recettori di riserva.
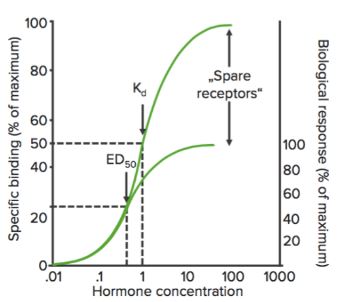
I recettori di riserva, almeno teoricamente, aumentano la sensibilità del farmaco.
Tossicità, curve di tossicità e rapporto terapeutico
Gli effetti avversi di un farmaco sono solitamente dose-dipendenti. Per studiare l’efficacia e la tossicità del farmaco, i suoi effetti sono testati in una popolazione e tracciati contro la concentrazione o la dose. Si ottengono curve sigmoidali (se si prendono le concentrazioni logiche sull’asse X), che possono riflettere gli effetti osservati.
Per esempio, nella figura seguente, un agente sedativo/ipnotico mostra l’azione sedativa ad un certo intervallo di concentrazione e la morte oltre tale intervallo. La dose (o la concentrazione) alla quale il farmaco mostra l’effetto desiderato (sedazione) nel 50% della popolazione è ED50.
La dose (o la concentrazione) alla quale il farmaco mostra gli effetti tossici (per esempio, depressione respiratoria) nel 50% della popolazione è TD50. La dose (o la concentrazione) alla quale il farmaco mostra l’effetto letale (morte) nel 50% della popolazione è LD50. (Questo è usato per gli esperimenti sugli animali.)
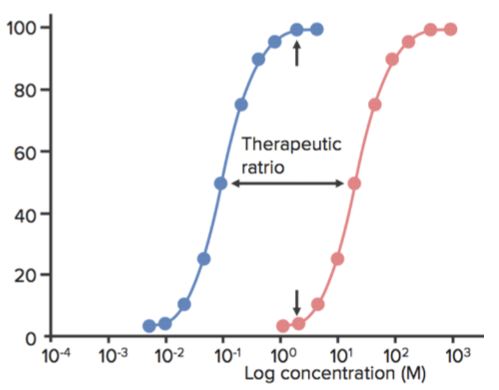
La finestra terapeutica è l’intervallo di dose tra la minima concentrazione efficace e la minima concentrazione tossica. L’indice terapeutico (TI) è definito come il rapporto tra il TD50 e l’ED50 o TI=TD50/ED50.
- Questo è talvolta chiamato anche rapporto terapeutico.
- Quando un farmaco ha un basso TI, significa che aumentando la sua dose può facilmente produrre effetti tossici (o letali).
- Gli antidepressivi triciclici e il litio, per esempio, hanno un basso TI, e la loro dose deve, quindi, essere monitorata molto attentamente nei pazienti. L’imipramina, per esempio, può essere fatale se somministrata in 5-6 volte la dose massima giornaliera.
- Più alto è il TI, più sicuro è il farmaco (l’esempio è la penicillina).
Studiare per la scuola medica e gli esami con Lecturio.
- USMLE Step 1
- USMLE Step 2
- COMLEX Level 1
- COMLEX Level 2
- ENARM
- NEET