Stabilitet hos karbokationsintermediärer
Vi vet att det hastighetsbegränsande steget i en SN1-reaktion är det första – bildandet av karbokationsintermediären. Hastigheten för detta steg – och därmed hastigheten för den totala substitutionsreaktionen – beror på aktiveringsenergin för den process där bindningen mellan kolet och den avgående gruppen bryts och en karbokation bildas. Enligt Hammonds postulat (avsnitt 6.2B) gäller att ju stabilare karbokationsintermediären är, desto snabbare kommer detta första bindningsbrytande steg att inträffa. Med andra ord beror sannolikheten för att en nukleofil substitutionsreaktion ska gå via en dissociativ (SN1) mekanism i hög grad på stabiliteten hos den karbokationsintermediär som bildas.
Den kritiska frågan blir nu vad som stabiliserar en karbokation.
Om det krävs en elektronåterkallande grupp för att stabilisera en negativ laddning, vad stabiliserar då en positiv laddning? En elektronavgivande grupp!
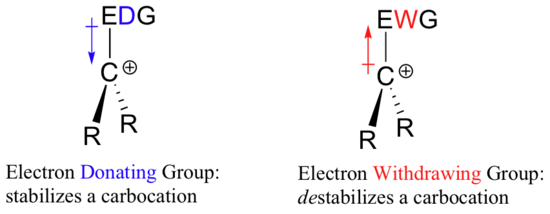
En positivt laddad art som en karbokation är mycket elektronfattig, och därför kommer allt som donerar elektrontäthet till centrumet för elektronfattigdomen att hjälpa till att stabilisera den. Omvänt kommer en karbokation att destabiliseras av en grupp som drar till sig elektroner.
Alkylgrupper – metyl, etyl och liknande – är svaga elektrondonatorgrupper och stabiliserar därför närliggande karbokationer. Detta innebär att mer substituerade karbokationer i allmänhet är stabilare: en tert-butylkarbokation är till exempel stabilare än en isopropylkarbokation. Primära karbokationer är mycket instabila och observeras inte ofta som reaktionsintermediärer; metylkarbokationer är ännu mindre stabila.
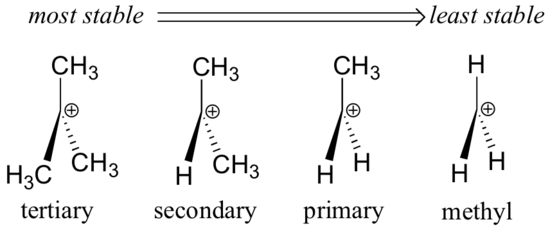
Alkylgrupper är elektrondonatorer och karbokationsstabiliserande eftersom elektronerna kring de närliggande kolvätena dras till den närliggande positiva laddningen, och på så sätt minskar elektronfattigdomen hos det positivt laddade kolet något.
Det är dock inte korrekt att säga att karbokationer med högre substitution alltid är mer stabila än de med mindre substitution. Precis som elektronavgivande grupper kan stabilisera en karbokation, verkar elektronavdragande grupper för att destabilisera karbokationer. Karbonylgrupper är elektronåterkallande genom induktiva effekter på grund av polariteten hos C=O-dubbelbindningen. Det är möjligt att visa i laboratoriet (se avsnitt 16.1D) att karbokation A nedan är stabilare än karbokation B, trots att A är en primär karbokation och B är sekundär.
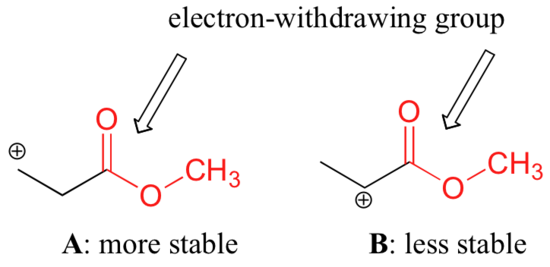
Den olika stabiliteten kan förklaras genom att beakta esterkarbonylens elektrondragande induktiva effekt. Minns att induktiva effekter – oavsett om de är elektronindragande eller donerande – förmedlas genom kovalenta bindningar och att effektens styrka minskar snabbt när antalet mellanliggande bindningar ökar. Med andra ord minskar effekten med avståndet. I art B är den positiva laddningen närmare karbonylgruppen, vilket innebär att den destabiliserande elektron-återkallande effekten är starkare än i art A.
I nästa kapitel kommer vi att se hur den karbokationsdestabiliserande effekten av elektron-återkallande fluorsubstituenter kan användas i experiment som är utformade för att besvara frågan om huruvida en biokemisk nukleofil substitutionsreaktion är SN1 eller SN2.
Stabilisering av en karbokation kan också ske genom resonanseffekter, och som vi redan har diskuterat i syra-baskapitlet är resonanseffekter som regel kraftfullare än induktiva effekter. Betrakta det enkla fallet med en bensylisk karbokation:
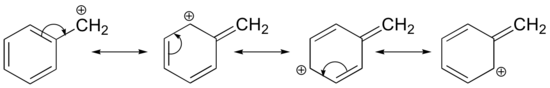
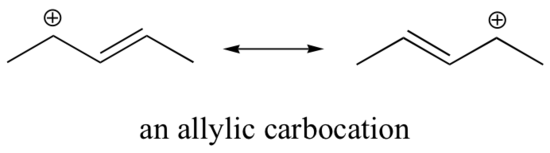
Denna karbokation är jämförelsevis stabil. I det här fallet är elektrondonation en resonanseffekt. Tre ytterligare resonansstrukturer kan ritas för denna karbokation där den positiva laddningen är placerad på en av tre aromatiska kolväten. Den positiva laddningen är inte isolerad på det bensyliska kolet, utan den är snarare delokaliserad runt den aromatiska strukturen: denna delokalisering av laddningen resulterar i en betydande stabilisering. Som ett resultat av detta är bensyliska och allyliska karbokationer (där det positivt laddade kolet är konjugerat till en eller flera icke-aromatiska dubbelbindningar) betydligt stabilare än till och med tertiära alkylkarbokationer.
Då heteroatomer som syre och kväve är mer elektronegativa än kol skulle man kunna förvänta sig att de per definition skulle vara elektronåterkallande grupper som destabiliserar karbokationer. I själva verket är det ofta tvärtom: om syre- eller kväveatomen befinner sig i rätt position är den övergripande effekten en karbokationsstabilisering. Detta beror på det faktum att även om dessa heteroatomer är elektronuttagande grupper genom induktion är de elektronavgivande grupper genom resonans, och det är denna resonanseffekt som är kraftfullare. (Vi stötte tidigare på samma idé när vi betraktade den relativa surheten och basiskheten hos fenoler och aromatiska aminer i avsnitt 7.4). Betrakta de två paren av karbokationsarter nedan:
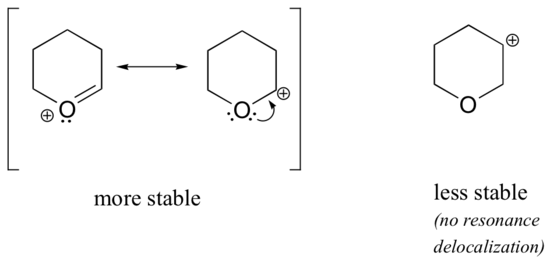
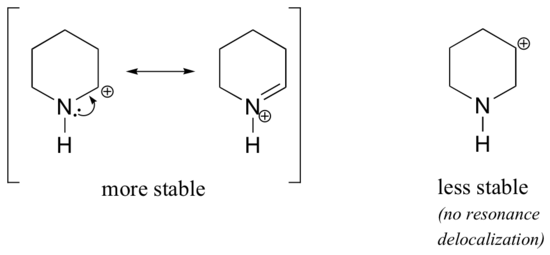
I de mer stabila karbokationerna fungerar heteroatomen som en elektrondonatorisk grupp genom resonans: i själva verket är det ensamma paret på heteroatomen tillgängligt för att delokalisera den positiva laddningen. I de mindre stabila karbokationerna är det positivt laddade kolet mer än en bindning bort från heteroatomen, och därmed är inga resonanseffekter möjliga. I dessa karbokationsarter destabiliserar heteroatomerna faktiskt den positiva laddningen, eftersom de genom induktion drar tillbaka elektroner.
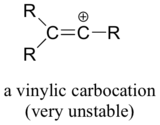
Finalt är vinyliska karbokationer, där den positiva laddningen sitter på ett dubbelbundet kol, mycket instabila och därmed osannolikt att de bildas som intermediärer i någon reaktion.
Exempel 7.9.1
I vilken av nedanstående strukturer förväntas karbokationen vara mer stabil? Förklara.
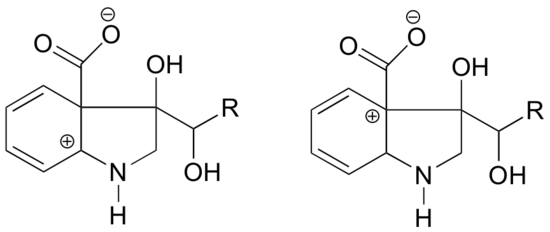
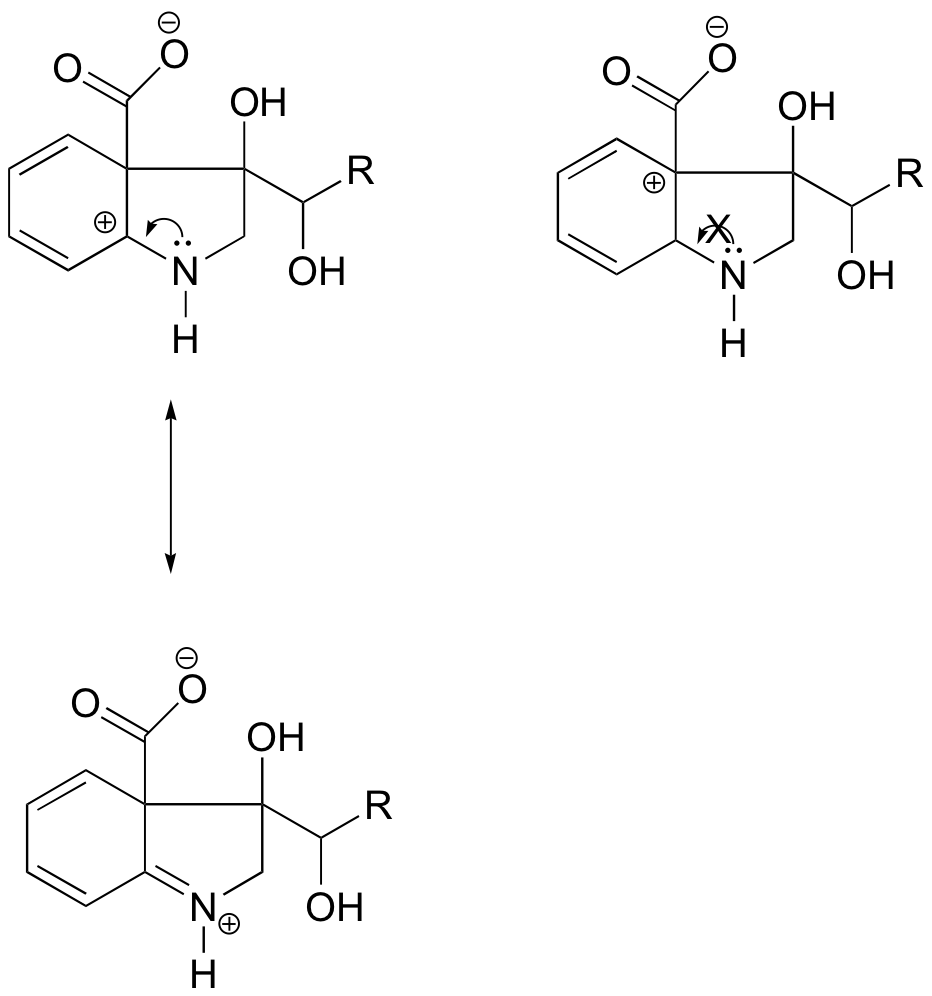
Svar
I karbokationen till vänster är den positiva laddningen placerad i en sådan position i förhållande till kvävet att det ensamma elektronparet på kvävet kan doneras för att fylla den tomma banan. Detta är inte möjligt för karbokationsarten till höger.
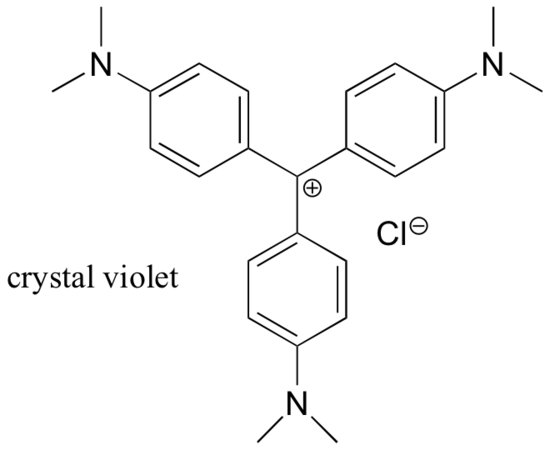
För det mesta är karbokationer mycket energirika, övergående intermediära arter i organiska reaktioner. Det finns dock några ovanliga exempel på mycket stabila karbokationer som tar formen av organiska salter. Kristallviolett är det vanliga namnet på kloridsaltet av den karbokation vars struktur visas nedan. Lägg märke till de strukturella möjligheterna till omfattande resonansdelokalisering av den positiva laddningen och närvaron av tre elektrondonerande amingrupper.
Exempel 7.9.2
Teckna en resonansstruktur för kristallviolettkatjonen där den positiva laddningen är delokaliserad till en av kväveatomerna.
Svar 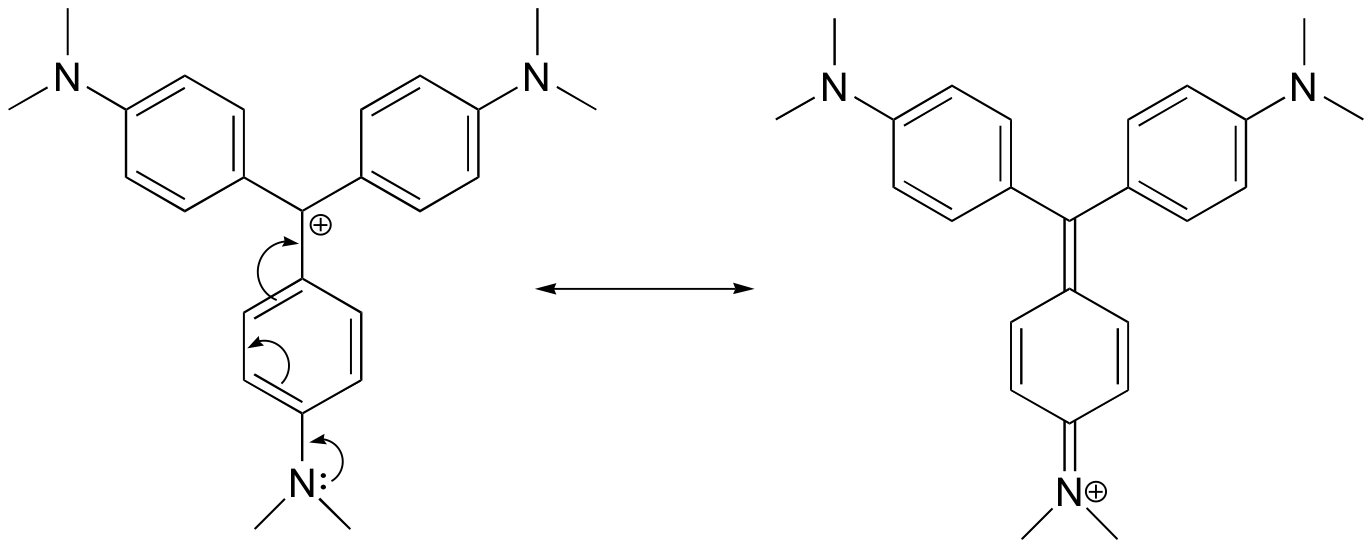
När man överväger möjligheten att en nukleofil substitutionsreaktion går via en SN1-väg är det viktigt att utvärdera stabiliteten hos den hypotetiska karbokationsintermediären. Om denna intermediär inte är tillräckligt stabil måste en SN1-mekanism anses osannolik, och reaktionen går troligen via en SN2-mekanism. I nästa kapitel kommer vi att se flera exempel på biologiskt viktiga SN1-reaktioner där den positivt laddade intermediären stabiliseras av induktiva och resonanseffekter som är inneboende i dess egen molekylära struktur.
Exempel 7.9.3
Ange vilken karbokation i varje par nedan som är stabilast, eller om de förväntas vara ungefär lika stabila. Förklara ditt resonemang.
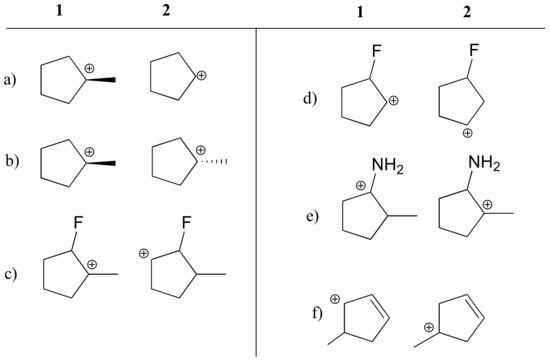
Svar
a) 1 (tertiär vs. sekundär karbokation)
b) lika
c) 1 (tertiär vs. sekundär karbokation)
b) lika
c) 1 (tertiär vs. sekundär karbokation). sekundär karbokation)
d) 2 (positiv laddning är längre från elektronåterkallande fluor)
e) 1 (ensam par på kväve kan donera elektroner genom resonans)
f) 1 (allylisk karbokation – positiv laddning kan flyttas till ett andra kol)
f) 1 (allylisk karbokation – positiv laddning kan flyttas till ett andra kol)