Stabilność pośrednich karbokationów
Wiemy, że etapem ograniczającym szybkość reakcji SN1 jest pierwszy etap – tworzenie tego pośredniego karbokationu. Szybkość tego etapu – a tym samym szybkość całej reakcji substytucji – zależy od energii aktywacji procesu, w którym następuje rozerwanie wiązania między węglem a grupą opuszczającą i utworzenie karbokationu. Zgodnie z postulatem Hammonda (rozdział 6.2B), im bardziej stabilny jest pośredni karbokation, tym szybciej nastąpi ten pierwszy etap zerwania wiązania. Innymi słowy, prawdopodobieństwo reakcji substytucji nukleofilowej przebiegającej według mechanizmu dysocjacyjnego (SN1) zależy w dużym stopniu od stabilności tworzącego się pośredniego karbokationu.
Krytycznym pytaniem staje się teraz, co stabilizuje karbokation?
So jeśli to wymaga wycofania elektronu grupa do stabilizacji ładunek ujemny, co będzie stabilizować ładunek dodatni? Grupa oddająca elektrony!
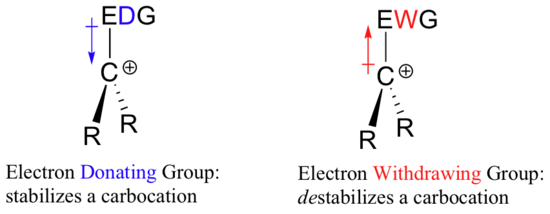
Dodatnio naładowany gatunek, taki jak karbokation, jest bardzo ubogi w elektrony, a zatem wszystko, co oddaje gęstość elektronową do centrum ubóstwa elektronowego, pomoże go ustabilizować. I odwrotnie, karbokation będzie destabilizowany przez grupę wycofującą elektrony.
Grupy alkilowe – metylowe, etylowe i podobne – są słabymi grupami oddającymi elektrony, a zatem stabilizują pobliskie karbokationy. Oznacza to, że ogólnie rzecz biorąc, bardziej podstawione karbokationy są bardziej stabilne: na przykład karbokation tert-butylowy jest bardziej stabilny niż karbokation izopropylowy. Karbokationy pierwszorzędowe są wysoce niestabilne i nie są często obserwowane jako intermediaty reakcji; karbokationy metylowe są jeszcze mniej stabilne.
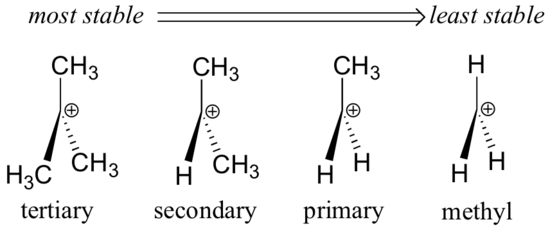
Grupy alkilowe są oddające elektrony i stabilizujące karbokationy, ponieważ elektrony wokół sąsiednich węgli są przyciągane w kierunku pobliskiego ładunku dodatniego, zmniejszając w ten sposób nieznacznie ubóstwo elektronowe dodatnio naładowanego węgla.
Nie jest jednak dokładne stwierdzenie, że karbokationy z większą ilością podstawników są zawsze bardziej stabilne niż te z mniejszą ilością podstawników. Tak jak grupy oddające elektrony mogą stabilizować karbokationy, grupy odbierające elektrony działają destabilizująco na karbokationy. Grupy karbonylowe oddają elektrony poprzez efekty indukcyjne, ze względu na polarność wiązania podwójnego C=O. Możliwe jest wykazanie w laboratorium (patrz sekcja 16.1D), że karbokation A poniżej jest bardziej stabilny niż karbokation B, mimo że A jest karbokationem pierwszorzędowym, a B drugorzędowym.
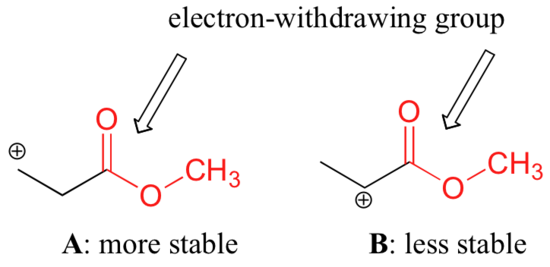
Różnica w stabilności może być wyjaśniona przez rozważenie efektu indukcyjnego polegającego na odbieraniu elektronów przez karbonyl estrowy. Przypomnijmy, że efekty indukcyjne – czy to oddawania czy odbierania elektronów – są przekazywane przez wiązania kowalencyjne i że siła efektu szybko maleje wraz ze wzrostem liczby wiązań pośrednich. Innymi słowy, efekt maleje wraz z odległością. W gatunku B ładunek dodatni jest bliżej grupy karbonylowej, a więc destabilizujący efekt wycofania elektronu jest silniejszy niż w gatunku A.
W następnym rozdziale zobaczymy, jak destabilizujący efekt karbokationów wywołany przez wycofujące elektrony podstawniki fluorowe może być wykorzystany w eksperymentach mających na celu odpowiedź na pytanie, czy biochemiczna reakcja substytucji nukleofilowej jest reakcją SN1 czy SN2.
Stabilizacja karbokationu może również zachodzić poprzez efekty rezonansowe, a jak już omawialiśmy w rozdziale kwasowo-zasadowym, efekty rezonansowe z reguły są silniejsze niż efekty indukcyjne. Rozważmy prosty przypadek karbokationu benzylowego:
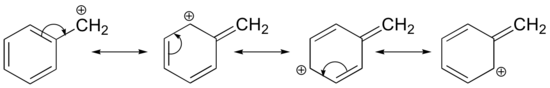
Ten karbokation jest stosunkowo stabilny. W tym przypadku oddawanie elektronów jest efektem rezonansowym. Można narysować trzy dodatkowe struktury rezonansowe dla tej karbokationu, w których ładunek dodatni jest zlokalizowany na jednym z trzech aromatycznych węgli. Ładunek dodatni nie jest izolowany na węglu benzylowym, lecz jest delokalizowany wokół struktury aromatycznej: ta delokalizacja ładunku powoduje znaczną stabilizację. W rezultacie, karbokationy benzylowe i allilowe (gdzie dodatnio naładowany węgiel jest sprzężony z jednym lub więcej niearomatycznymi wiązaniami podwójnymi) są znacznie bardziej stabilne niż nawet karbokationy trzeciorzędowych alkilów.
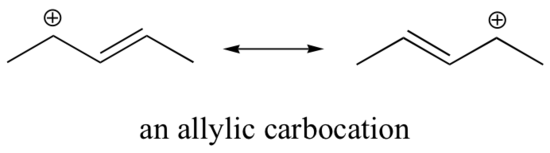
Ponieważ heteroatomy takie jak tlen i azot są bardziej elektronegatywne niż węgiel, można by się spodziewać, że z definicji będą one grupami wycofującymi elektrony, które destabilizują karbokationy. W rzeczywistości, często jest odwrotnie: jeśli atom tlenu lub azotu znajduje się w odpowiedniej pozycji, ogólnym efektem jest stabilizacja karbokationów. Wynika to z faktu, że chociaż heteroatomy te są grupami odbierającymi elektrony przez indukcję, są one grupami oddającymi elektrony przez rezonans, i to właśnie ten efekt rezonansu jest silniejszy. (Z tą samą ideą zetknęliśmy się wcześniej, rozważając względną kwasowość i zasadowość fenoli i amin aromatycznych w rozdziale 7.4). Rozważmy dwie pary karbokationów poniżej:
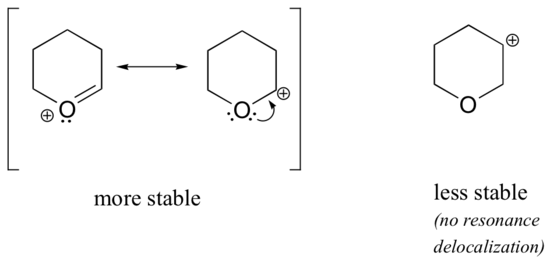
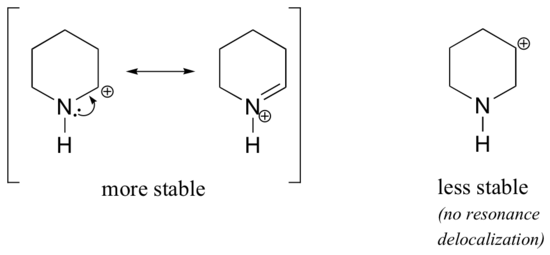
W bardziej stabilnych karbokationach, heteroatom działa jak grupa oddająca elektrony poprzez rezonans: w efekcie, samotna para na heteroatomie jest dostępna do delokalizacji ładunku dodatniego. W mniej stabilnych karbokationach dodatnio naładowany węgiel jest oddalony o więcej niż jedno wiązanie od heteroatomu, a zatem efekty rezonansowe nie są możliwe. W rzeczywistości, w tych gatunkach karbokationów heteroatomy faktycznie destabilizują ładunek dodatni, ponieważ są one wycofujące elektrony przez indukcję.
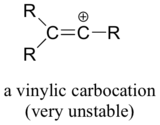
Wreszcie, karbokationy winylowe, w których ładunek dodatni znajduje się na podwójnie związanym węglu, są bardzo niestabilne i dlatego mało prawdopodobne jest, aby tworzyły się jako intermediaty w jakiejkolwiek reakcji.
Przykład 7.9.1
W której z poniższych struktur karbokation powinien być bardziej stabilny? Wyjaśnij.
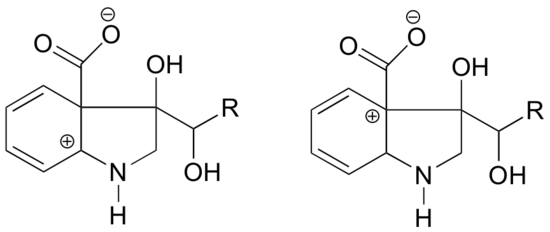
Odpowiedź
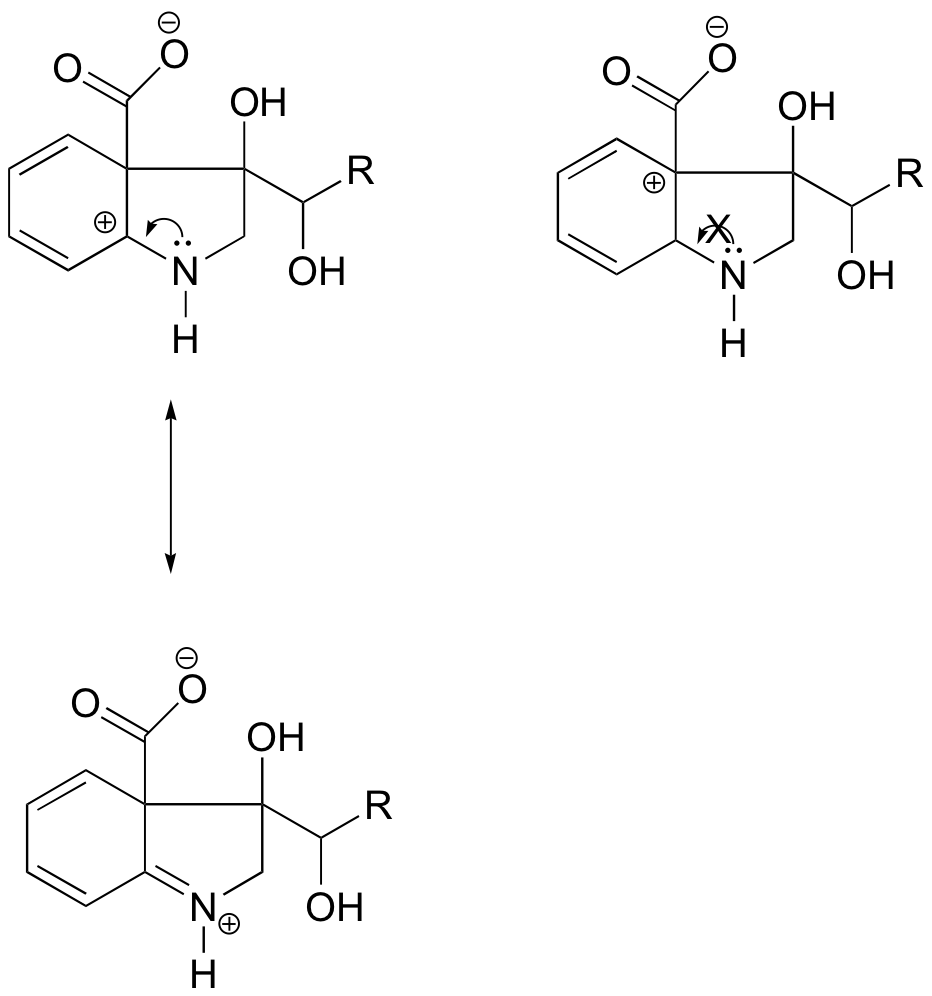
W karbokationie po lewej stronie ładunek dodatni znajduje się w takim położeniu względem azotu, że samotna para elektronów na azocie może zostać przekazana, aby wypełnić pusty orbital. Nie jest to możliwe w przypadku karbokationów po prawej stronie.
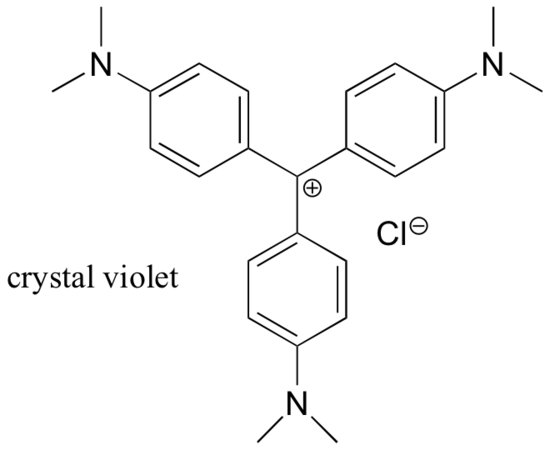
W przeważającej części, karbokationy są bardzo wysokoenergetycznymi, przejściowymi gatunkami pośrednimi w reakcjach organicznych. Istnieją jednak pewne niezwykłe przykłady bardzo stabilnych karbokationów, które przyjmują postać soli organicznych. Fiolet krystaliczny to potoczna nazwa soli chlorkowej karbokationu, którego struktura przedstawiona jest poniżej. Zwróć uwagę na strukturalne możliwości szerokiej rezonansowej delokalizacji ładunku dodatniego oraz obecność trzech elektronodonujących grup aminowych.
Przykład 7.9.2
Narysuj strukturę rezonansową kationu fioletu krystalicznego, w której ładunek dodatni jest delokalizowany do jednego z atomów azotu.
Odpowiedź 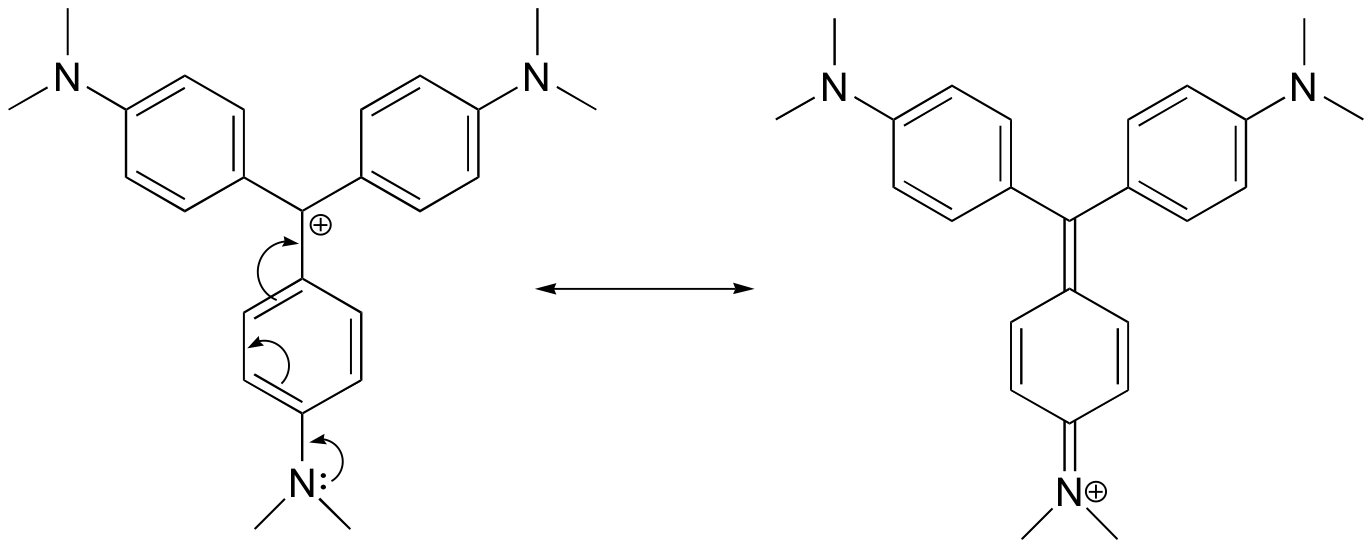
Przy rozważaniu możliwości, że reakcja substytucji nukleofilowej przebiega na drodze SN1, krytyczna jest ocena stabilności hipotetycznego pośredniego karbokationu. Jeśli ten półprodukt nie jest wystarczająco stabilny, mechanizm SN1 musi być uznany za mało prawdopodobny, a reakcja prawdopodobnie przebiega według mechanizmu SN2. W następnym rozdziale zobaczymy kilka przykładów biologicznie ważnych reakcji SN1, w których dodatnio naładowany element pośredni jest stabilizowany przez efekty indukcyjne i rezonansowe właściwe dla jego własnej struktury molekularnej.
Przykład 7.9.3
Powiedz, która karbokation w każdej poniższej parze jest bardziej stabilna, lub czy oczekuje się, że będą one w przybliżeniu równe. Wyjaśnij swoje rozumowanie.
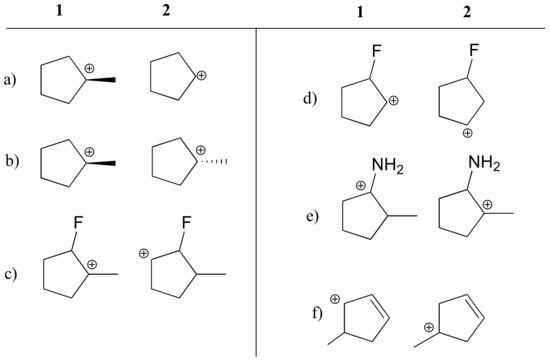
Odpowiedź
a) 1 (karbokation trzeciorzędowy vs. karbokation drugorzędowy)
b) równe
c) 1 (karbokation trzeciorzędowy vs. drugorzędowa karbokacja)
d) 2 (ładunek dodatni jest dalej od odbierającego elektrony fluoru)
e) 1 (samotna para na azocie może oddać elektrony przez rezonans)
f) 1 (karbokacja allilowa – ładunek dodatni może być delokalizowany na drugi węgiel)
e) 1 (karbokacja allilowa – ładunek dodatni może być delokalizowany na drugi węgiel)
.