Renální transport cystinu
Aminokyseliny se snadno filtrují v glomerulu a procházejí téměř úplnou reabsorpcí v proximálních tubulárních buňkách. Pouze 0,4 % filtrovaného cystinu se objeví v moči. Různí autoři studovali transport aminokyselin v buněčných membránách získaných z proximálního tubulu ledvin lidí, potkanů a králíků. Za reabsorpci cystinu jsou zodpovědné nejméně 2 transportní systémy:
-
Vysokoafinitní systém: Tento systém je postižen u osob s cystinurií. Vysoce afinitní systém zprostředkovává vychytávání 10 % L-cystinu a dibazických aminokyselin na apikální membráně přímého třetího segmentu (S3) proximálního tubulu.
-
Nízko afinitní systém: Tento systém je přítomen v části S1-S2 proximálního tubulu a je zodpovědný za 90 % reabsorpce L-cystinu. Proces nízké afinity zvyšuje proces vysoké afinity. Po absorpci je každá molekula cystinu intracelulárně přeměněna na 2 molekuly cysteinu. Cystein opouští bazolaterální membránu.
Genetické studie DNA z rodin s cystinurií odhalily defektní gen umístěný na chromozomu 2. Cystinurie se vyskytuje na chromozomu 2, kde se vyskytuje i cystin. Gen, který kóduje cystinový transportér, původně označovaný jako rBAT, je nyní v mezinárodní databázi genomů znám jako SLC3A1 (SLC jako solute carrier). Druhý gen pro cystinurii na chromozomu 19 se nazývá SLC7A9. Normální gen SLC7A9 kóduje podjednotku cystinového transportéru zvanou b 0,+ AT (aminokyselinový transportér). Proces vychytávání cystinu je aktivován produkty genů SLC3A1 a SLC7A9. Transport L-cystinu ve vezikulech kartáčové membrány je nezávislý na sodíku a elektrogenní. U osob s cystinurií není pohyb cystinu nebo cysteinu z tubulárních buněk do krve ovlivněn.
Střevní transport cystinu
Vysokoafinitní transportér je přítomen v apikální membráně kartáčové hranice jejuna a je zodpovědný za absorpci cystinu a dibazických aminokyselin. Většina pacientů s cystinurií má poruchu vstřebávání cystinu; nedostatek cystinu však není klinicky významný, protože vstřebávání aminokyselin s krátkým řetězcem není ovlivněno.
Normálně jsou cystin a ostatní dibazické aminokyseliny (tj. ornitin, lysin, arginin) filtrovány v glomerulu a reabsorbovány v proximálním stočeném tubulu pomocí vysokoafinitního luminálního transmembránového kanálu. Defekty tohoto kanálu způsobují zvýšenou sekreci dibazických aminokyselin v moči. Zatímco ornitin, lysin a arginin jsou zcela rozpustné, cystin je při fyziologických hodnotách pH moči 5-7 relativně nerozpustný, s pKa na úrovni 8,3. Při úrovni pH moči 7,8 a 8 je příslušná rozpustnost cystinu téměř dvojnásobná a trojnásobná.
Dent a Senior prokázali, že rozpustnost cystinu je závislá na pH. Rozpustnost cystinu v moči je přibližně 250 mg/l (1 mmol/l) do hodnoty pH 7, ale rozpustnost se zvyšuje s vyšší hodnotou pH až o 500 mg/l (2 mmol/l) nebo více nad hodnotu pH 7,5, jak je znázorněno na obrázku níže. Měření v moči jasně ukázala, že rozpustnost cystinu se lineárně zvyšuje se zvyšující se iontovou silou. Pak a jeho kolegové prokázali, že při zvýšení iontové síly z 0,005-0,3 lze v každém litru roztoku rozpustit přibližně 70 mg dalšího cystinu. Kromě toho se při stejné iontové síle a pH rozpustnost cystinu liší v závislosti na konkrétním typu přítomného elektrolytu. Viz obrázek níže.
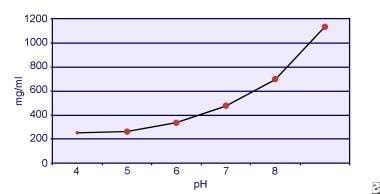 Rozpustnost cystinu v moči.
Rozpustnost cystinu v moči. Pokusy in vitro provedené Pakem a Fullerem v roce 1983 ukázaly, že nejvyšší rozpustnosti je dosaženo v přítomnosti chloridu vápenatého, následovaného chloridem hořečnatým a sodným. Kromě toho rozpustnost cystinu ovlivňují také makromolekuly v moči. Bylo prokázáno, že přítomnost koloidů v normální moči zvyšuje rozpustnost cystinu; mechanismus tohoto působení však není jasný. Protože krystalizaci cystinu nic nebrání, je hlavním určujícím faktorem přesycení moči. U jedinců s cystinurií nedochází k heterogenní nukleaci kalcium oxalátu, brushitu nebo hydroxyapatitu.
Rizikové faktory pro krystalizaci cystinu zahrnují (1) nízkou hladinu pH, (2) sníženou iontovou sílu, (3) přítomnost krystalů cystinu a (4) nízkou hladinu makromolekul v moči.
Cystin je disulfidicky vázaný homodimer cysteinu a má následující strukturu:
COOH-CHNH2-CH2 -S-S-CH2-CHNH2- COOH Cystin
COOH-CHNH2-CH2 -SH Cystein
Cystin se vstřebává v tenkém střevě podobně jako v ledvinách. U osob s cystinurií je střevní absorpce cystinu také v různé míře narušena. Dalším zdrojem cystinu v séru je metabolismus methioninu. Na transportu aminokyselin přes plazmatickou membránu se podílejí dvě domény membránových glykoproteinů typu II. První je rBAT a druhou je 4F2HC (těžký řetězec antigenu 4F2)
Dvě třetiny osob s cystinurií, kterým se tvoří kameny, mají čistě cystinové kameny a jedna třetina má směs cystinových a kalcium oxalátových kamenů. V roce 2002 Martins et al. uvedli, že k precipitaci oxalátu vápenatého dochází spíše procesem salting-out, tj. snížením rozpustnosti látky v důsledku přidání jiné látky do systému, než procesem heterogenní nukleace. Hypocitraturie, hyperkalciurie a hyperurikosurie jsou také často spojeny s cystinurií. Vzhledem k relativně homogenní krystalické struktuře bez lamelových štěpných rovin patří čisté cystinové kameny k nejtvrdším na Dretlerově indexu křehkosti kamenů.
Cystinurie je autozomálně recesivní onemocnění, které se dělí na 3 podtypy: Rosenberg I, II a III. Cystinurie typu I je nejčastější variantou a byla zmapována do pásma 2p16.3. Heterozygoti typu I vykazují normální aminoacidurii. Klasická cystinurie, typy II a III, byly považovány za alelické varianty, ale vazebné analýzy odhalily, že u typu III jde o defekt necharakterizovaného genu (SLC7A9) v pásu 19q13.1. V případě typu III se jedná o defekt genu SLC7A9. Heterozygoti typů II a III mají často cystinurii bez cystinových kamenů a mohou mít zvýšené riziko jiných typů urolitiázy. Heterozygoti typu I se vyznačují normální hladinou cystinu v moči.
Na rozdíl od homozygotů typu I a II vykazují homozygoti typu III zvýšení plazmatické koncentrace cystinu po perorálním podání cystinu. Harris et al popsali komplexní povahu genetiky cystinurie měřením hladiny vylučování cystinu močí u rodičů (obligátních heterozygotů) probandů s cystinurií a zjistili plně recesivní alely (oba rodiče vylučovali cystin v referenčním rozmezí) a dominantní alely (oba rodiče vylučovali cystin ve vysokých hladinách).
K klinické klasifikaci cystinurie lze u každého z rodičů probandů stanovit fenotyp I (recesivní, hladina cystinu v moči < 100 µmol/g kreatininu), fenotyp II (dominantní, hladina cystinu v moči >1000 µmol/g kreatininu) a fenotyp III (částečně dominantní, hladina cystinu v moči 100-1000 µmol/g kreatininu). Cystinurii lze také klasifikovat na základě věku, ve kterém se poprvé objeví příznaky (tj. dětský, juvenilní, adolescentní).
U zdravých jedinců je horní hranice vylučování cystinu 20 mg/g kreatininu (< 10 µmol/mmol kreatininu). Homozygoti vylučují více než 400 mg/d (1,7 mmol/d) a vylučování cystinu u homozygotů je obvykle 600-1400 mg/d (2,5-5,8 mmol/d). Heterozygoti s cystinurií typu I a III vylučují méně než 200 mg/d (0,8 mmol/d) a netvoří kameny. Heterozygoti s typem II vylučují až 200-400 mg/d, ale tito pacienti mohou tvořit kameny. Výskyt tvorby kamenů se zvyšuje, když koncentrace cystinu v moči překročí 700 µmol/l (170 mg/l).
Genetika
V posledních letech se s rozvojem molekulární biologie nashromáždily nové poznatky o patofyziologii cystinurie. V roce 1992 několik badatelů oznámilo klonování exprese 2,3kilobázové renální cDNA (D2H nebo rBAT), která indukovala na sodíku nezávislé vychytávání cystinu a dibazických aminokyselin v oocytech Xenopus laevis injikovaných cRNA. Gen rBAT byl mapován na chromozomu 2 (pás 2p21) mezi D2S119 a D2S288. Tento gen je nyní v databázi genomů pojmenován SLC3A1.
Imunohistochemické a in situ hybridizační studie odhalily, že rBAT je exprimován v buňkách segmentu S3 (pars recta) proximálního tubulu a tenkého střeva na luminální membráně kartáčové hranice. V roce 1995 Gasparini et al uvedli, že mutace v SLC3A1 se vyskytují u pacientů s cystinurií typu I a nikoli u pacientů s cystinurií typu II nebo III. Do dnešního dne bylo popsáno více než 160 různých mutací, včetně malých i velkých delecí párů bází DNA z genu. Jedna z nejčastějších genetických změn v SLC3A1 se nazývá M467T a většina mutací bývá populačně specifická. Mutace M467T je poměrně častá u středomořské populace. Zajímavé je, že ve španělské kohortě rodin tvořila 40 % mutací a u pacientů studovaných v kanadském Quebecu byla vzácná.
V roce 1999 byl izolován gen SLC7A9 (BAT1). Gen kóduje protein o 487 aminokyselinách a byl mapován na chromozomu 19 (pás 19q13) mezi D19S414 a D19S220. Zdá se, že produkt BAT1 je membránový protein s 12 membránovými oblastmi. Mutace v genu BAT1 pravděpodobně způsobují cystinurii jiného než I. typu (Rosenbergův typ II a III). Mutace v lokusu 19q jsou zvláště časté u libyjských Židů a riziko vzniku močových kamenů u pacientů, kteří zdědí 2 takové mutace v lokusu 19q, je zhruba srovnatelné s rizikem u pacientů, kteří zdědí 2 mutace v rBAT.
Bylo zaznamenáno 116 mutací v tomto genu. Nejčastější mutace u libyjských Židů vedla k tomu, že v proteinu nahradil aminokyselinový zbytek valin methionin (V170M). U heterozygotů s mutací V170M se koncentrace cystinu v moči pohybují v rozmezí 86-1238 µmol/g kreatininu. Některé hodnoty u heterozygotů s mutací V170M tedy odpovídají cystinurii III. typu a jiné cystinurii II. typu.
Zjevným rozlišovacím znakem mezi cystinurií II. a III. typu je absence střevní absorpce cystinu u homozygotů II. typu. V roce 2000 navrhl Pras novou klasifikaci založenou na molekulární analýze. Nedávno Dello Strologo et al navrhli novou genetickou klasifikaci, a to následující:
-
Typ A, mutace obou alel SLC3A1: Heterozygoti vykazují normální vylučování aminokyselin močí.
-
Typ B, mutace obou alel SLC7A9: Heterozygoti obvykle vykazují zvýšené vylučování cystinu a dibazických aminokyselin močí.
-
Typ AB, cystinurie způsobená 1 mutací v SLC3A1 a 1 mutací v SLC7A9: Cystinurie smíšeného typu může být způsobena interakcí 2 různých mutovaných genů a protein kódovaný genem 19q přímo interaguje s rBAT v segmentu S3 proximálního tubulu (viz tabulka).
Martens et al (2008) nedávno uvedli 3 syndromy s delecí genu spojené s cystinurií typu A: syndrom delece 2p21, syndrom hypotonie-cystinurie (HCS) a atypickou formu syndromu hypotonie-cystinurie. U pacientů s HCS chybí obě alely SLC3A1 a PREPL. U atypického HCS je odstraněn další gen (C2orf34).
Tabulka. Klasifikace cystinurie (Otevřít tabulku v novém okně)
|
Rosenberg a kol. |
Typ I |
Typ II |
Typ III |
|
Molekulární |
Typ I |
Není typ I |
|
|
Odpovědný gen |
SLC3A1 |
SLC7A9 |
|
|
Pásmo |
2p21 |
19q13.1 |
|
|
Č. mutací |
>60 |
||
|
Nejčastější mutace |
M467 |
V170M |
|
|
Populace postižená |
Středomořské španělské osoby, 40% |
Libijští Židé |
|
|
Míra delece |
54% |
25% |
|
|
Protein |
rBAT |
BAT1 |
|
|
Transportní systém aminokyselin |
|||
|
Lokalizace v proximálním přeměněném tubulu |
S3 |
S1, S2 |
|
|
Transportní charakteristika |
Vysoká afinita, nízká kapacita |
Nízká afinita, vysoká kapacita |
|
|
Klinické rysy |
|||
|
Homozygoti |
Symptomatičtí |
přibližně 90% symptomatických |
|
|
Heterozygoti |
Asymptomatičtí |
přibližně 10%-ní13% symptomatických |
|
|
Hladina cystinu v moči |
Normální |
Zvýšená +++++ |
Zvýšená + |
|
Hladina cystinu v plazmě. po orálním zátěžovém testu |
Stejná |
Stejná nebo mírně zvýšená |
Zvýšená |
|
Střevní transport |
Přítomná (nedochází k transportu cystinu, lysinu, nebo argininu) |
Absentní |
Redukovaný |
Nejnovější důkazy naznačují, že komplex 4F2HC/4F2LC představuje za transportní systém aminokyselin Y+L na bazolaterálním povrchu střevních a ledvinových proximálních tubulárních buněk a že mutace genu 4F2LC (SLC7A7) v pásmu 14q11-13 způsobuje vzácné recesivní onemocnění zvané intolerance lysinových bílkovin.
Shrnutí
rBAT, 90-kd glykoprotein typu II, je vysoce afinitní, na sodíku nezávislý transportér pro dibazické aminokyseliny v proximálních stočených ledvinových tubulech u hlodavců. lidský gen rBAT byl lokalizován na pásmu 2p21. Zajímavé je, že analýza vazeb naznačuje, že se jedná o stejnou oblast, ve které byl identifikován cystinurický lokus SLC3A1.U pacientů s cystinurií bylo celosvětově identifikováno 160 různých mutací v genu SLC3A1 a 116 v genu SLC7A9.
Cystinurie III. a II. typu (jiná než I. typu) byla spojena s pásem 19p13.1 (SLC7A9); k určení přesné úlohy genu SLC7A9 jsou však zapotřebí další studie. Přibližně u 50 % dětí se dvěma mutacemi SLC3A1 (klasická homozygotní cystinurie typu I) se během prvního desetiletí života objeví alespoň jeden kámen
.