Transporte renal de cistina
Los aminoácidos son fácilmente filtrados por el glomérulo y sufren una reabsorción casi completa por las células tubulares proximales. Sólo el 0,4% de la cistina filtrada aparece en la orina. Varios autores han estudiado el transporte de aminoácidos en membranas celulares obtenidas del túbulo renal proximal de humanos, ratas y conejos. Al menos dos sistemas de transporte son responsables de la reabsorción de cistina, a saber:
-
Sistema de alta afinidad: Este sistema está afectado en las personas con cistinuria. El sistema de alta afinidad media la captación del 10% de la L-cistina y los aminoácidos dibásicos en la membrana apical del tercer segmento recto (S3) del túbulo proximal.
-
Sistema de baja afinidad: Este sistema está presente en la parte S1-S2 del túbulo proximal y es responsable del 90% de la reabsorción de L-cistina. El proceso de baja afinidad aumenta el proceso de alta afinidad. Tras la absorción, cada molécula de cistina se convierte intracelularmente en 2 moléculas de cisteína. La cisteína sale por la membrana basolateral.
Los estudios genéticos del ADN de familias con cistinuria revelan un gen defectuoso localizado en el cromosoma 2. El gen que codifica el transportador de cistina, inicialmente denominado rBAT, se conoce ahora como SLC3A1 (SLC por solute carrier) en la base de datos internacional del genoma. Un segundo gen de la cistinuria en el cromosoma 19 se denomina SLC7A9. El gen SLC7A9 normal codifica una subunidad del transportador de cistina llamada b 0,+ AT (transportador de aminoácidos). El proceso de captación de cistina es activado por los productos de los genes SLC3A1 y SLC7A9. El transporte de L-cistina en la vesícula de la membrana del borde en cepillo es independiente del sodio y electrogénico. En las personas con cistinuria, el movimiento de cistina o cisteína desde las células tubulares a la sangre no está afectado.
Transporte intestinal de cistina
El transportador de alta afinidad está presente en la membrana apical del borde en cepillo del yeyuno y es responsable de la absorción de cistina y aminoácidos dibásicos. La mayoría de los pacientes con cistinuria tienen alterada la absorción de cistina; sin embargo, la deficiencia de cistina no es clínicamente significativa porque la absorción de aminoácidos de cadena corta no se ve afectada.
Normalmente, la cistina y los otros aminoácidos dibásicos (es decir, ornitina, lisina, arginina) se filtran en el glomérulo y se reabsorben en el túbulo contorneado proximal por un canal transmembrana luminal de alta afinidad. Los defectos en este canal provocan niveles elevados de secreción de aminoácidos dibásicos en la orina. Mientras que la ornitina, la lisina y la arginina son completamente solubles, la cistina es relativamente insoluble a niveles fisiológicos de pH urinario de 5-7, con un nivel de pKa de 8,3. A un nivel de pH urinario de 7,8 y 8, la solubilidad respectiva de la cistina casi se duplica y triplica.
Dent y Senior demostraron que la solubilidad de la cistina depende del pH. La solubilidad de la cistina en la orina es de aproximadamente 250 mg/L (1 mmol/L) hasta un nivel de pH de 7, pero la solubilidad aumenta con un nivel de pH más alto hasta 500 mg/L (2 mmol/L) o más por encima de un nivel de pH de 7,5, como se representa en la imagen siguiente. Las mediciones en la orina han demostrado claramente que la solubilidad de la cistina aumenta linealmente con el aumento de la fuerza iónica. Pak y sus colegas demostraron que se pueden disolver aproximadamente 70 mg de cistina adicional en cada litro de solución, con un aumento de la fuerza iónica de 0,005-0,3. Además, a la misma fuerza iónica y pH, la solubilidad de la cistina varía según el tipo particular de electrolito presente. Véase la imagen siguiente.
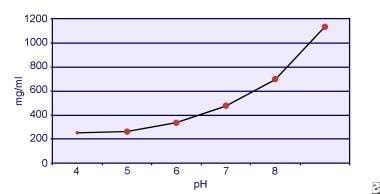 Solubilidad de la cistina en la orina.
Solubilidad de la cistina en la orina. Los experimentos in vitro realizados por Pak y Fuller en 1983 revelaron que la mayor solubilidad se logra en presencia de cloruro de calcio, seguido de cloruro de magnesio y de sodio. Además, la solubilidad de la cistina también se ve afectada por las macromoléculas urinarias. Se ha demostrado que la presencia de coloides en la orina normal aumenta la solubilidad de la cistina; sin embargo, el mecanismo de esta acción no está claro. Dado que nada inhibe la cristalización de la cistina, el principal determinante es la sobresaturación urinaria. La nucleación heterogénea de oxalato de calcio, brushita o hidroxiapatita no se produce en individuos con cistinuria.
Los factores de riesgo para la cristalización de cistina incluyen (1) un nivel de pH bajo, (2) una fuerza iónica reducida, (3) la presencia de cristales de cistina y (4) niveles bajos de macromoléculas urinarias.
La cistina es un homodímero de cisteína ligado a disulfuro y tiene la siguiente estructura:
COOH-CHNH2-CH2 -S-S-CH2-CHNH2- COOH Cistina
COOH-CHNH2-CH2 -SH Cisteína
La cistina se absorbe en el intestino delgado de forma similar a la de los riñones. En las personas con cistinuria, la absorción intestinal de la cistina también está alterada en diversos grados. El metabolismo de la metionina es otra fuente de cistina sérica. Se han implicado dos dominios de glicoproteínas de membrana de tipo II en el transporte de aminoácidos a través de la membrana plasmática. El primero es el rBAT, y el segundo es el 4F2HC (la cadena pesada del antígeno 4F2)
Dos tercios de las personas con cistinuria que forman cálculos hacen cálculos de cistina pura, y un tercio tienen una mezcla de cálculos de cistina y oxalato de calcio. En 2002, Martins et al informaron de que la precipitación de oxalato de calcio se produce por un proceso de salinización, es decir, la reducción de la solubilidad de una sustancia debido a la adición de otra sustancia al sistema, y no por el proceso de nucleación heterogénea. La hipocitraturia, la hipercalciuria y la hiperuricosuria también se asocian con frecuencia a la cistinuria. Dada su estructura cristalina relativamente uniforme sin planos de escisión laminar, los cálculos de cistina pura se encuentran entre los más duros en el índice de fragilidad de cálculos de Dretler.
La cistinuria es una enfermedad autosómica-recesiva dividida en 3 subtipos: Rosenberg I, II y III. La cistinuria tipo I es la variante más común y ha sido mapeada en la banda 2p16.3. Los heterocigotos del tipo I muestran una aminoaciduria normal. Se pensaba que la cistinuria clásica, tipos II y III, eran variantes alélicas, pero los análisis de vinculación han revelado que el tipo III es un defecto de un gen no caracterizado (SLC7A9) en la banda 19q13.1. Los heterocigotos de los tipos II y III suelen manifestar cistinuria sin cálculos de cistina y pueden tener un mayor riesgo de padecer otros tipos de urolitiasis. Los heterocigotos de tipo I se distinguen por los niveles normales de cistina en la orina.
A diferencia de los homocigotos de tipo I y II, los homocigotos de tipo III muestran un aumento de la concentración de cistina en plasma tras la administración de cistina por vía oral. Harris et al informaron de la naturaleza compleja de la genética de la cistinuria midiendo el nivel de excreción de cistina en la orina de los padres (heterocigotos obligados) de los probandos con cistinuria y encontraron alelos totalmente recesivos (ambos padres excretaban cistina en el rango de referencia) y alelos dominantes (ambos padres excretaban cistina a niveles elevados).
Para clasificar la cistinuria clínicamente, la cistina urinaria puede medirse en cada progenitor de un probando como fenotipo I (recesivo, nivel de cistina urinaria < 100 µmol/g de creatinina), fenotipo II (dominante, nivel de cistina urinaria >1000 µmol/g de creatinina) y fenotipo III (parcialmente dominante, nivel de cistina urinaria 100-1000 µmol/g de creatinina). La cistinuria también puede clasificarse en función de la edad de aparición de los síntomas (es decir, infantil, juvenil, adolescente).
En individuos sanos, el límite superior de excreción de cistina es de 20 mg/g de creatinina (< 10 µmol/mmol de creatinina). Los homocigotos excretan más de 400 mg/d (1,7 mmol/d), y la excreción de cistina en pacientes homocigotos suele ser de 600-1400 mg/d (2,5-5,8 mmol/d). Los heterocigotos con cistinuria de tipo I y III excretan menos de 200 mg/d (0,8 mmol/d) y no forman cálculos. Los heterocigotos de tipo II excretan hasta 200-400 mg/d, pero estos pacientes pueden formar cálculos. La incidencia de la formación de cálculos aumenta cuando la concentración de cistina en la orina supera los 700 µmol/L (170 mg/L).
Genética
En los últimos años, con los avances en biología molecular, se han acumulado nuevos conocimientos sobre la fisiopatología de la cistinuria. En 1992, varios investigadores informaron de la clonación de la expresión de un ADNc renal de 2,3 kilobases (D2H o rBAT) que inducía la captación independiente del sodio de la cistina y los aminoácidos dibásicos en oocitos Xenopus laevis inyectados con el ADNc. El gen rBAT fue mapeado en el cromosoma 2 (banda 2p21) entre D2S119 y D2S288. Este gen se denomina ahora SLC3A1 en la base de datos del genoma.
Los estudios inmunohistoquímicos y de hibridación in situ revelaron que rBAT se expresa en las células del segmento S3 (pars recta) del túbulo proximal y del intestino delgado en la membrana luminal del borde en cepillo. En 1995, Gasparini et al informaron de que las mutaciones en SLC3A1 se producían en pacientes con cistinuria de tipo I y no en pacientes con cistinuria de tipo II o III. Hasta la fecha, se han descrito más de 160 mutaciones diferentes, que incluyen tanto pequeñas como grandes deleciones de pares de bases de ADN del gen. Una de las alteraciones genéticas más comunes en SLC3A1 se denomina M467T, y la mayoría de las mutaciones tienden a ser específicas de la población. La mutación M467T es bastante común en las poblaciones mediterráneas. Curiosamente, representó el 40% de las mutaciones en una cohorte española de familias y fue rara en pacientes estudiados en Quebec, Canadá.
En 1999, se aisló el gen SLC7A9 (BAT1). El gen codifica una proteína de 487 aminoácidos y fue mapeado en el cromosoma 19 (banda 19q13) entre D19S414 y D19S220. El producto de la BAT1 parece ser una proteína de membrana con 12 regiones que la atraviesan. Las mutaciones en el gen BAT1 probablemente causan cistinuria no tipo I (Rosenberg tipo II y III). Las mutaciones en el locus 19q son especialmente comunes entre los judíos libios, y el riesgo de urolitiasis en los pacientes que heredan 2 de estas mutaciones del locus 19q es aproximadamente comparable al de los pacientes que heredan 2 mutaciones rBAT.
Se han descrito 116 mutaciones en este gen. La mutación más común en los judíos libios dio lugar a que una metionina sustituyera al residuo de aminoácido valina (V170M) en la proteína. En los heterocigotos con la mutación V170M, las concentraciones de cistina en la orina oscilan entre 86 y 1238 µmol/g de creatinina. Así, algunos de los valores en los heterocigotos V170M son consistentes con la cistinuria de tipo III y otros con la cistinuria de tipo II.
Una característica distintiva aparente entre la cistinuria de tipo II y la de tipo III es la falta de absorción intestinal de cistina en los homocigotos de tipo II. En 2000, Pras sugirió una nueva clasificación basada en el análisis molecular. Recientemente, Dello Strologo et al han propuesto una nueva clasificación genética, como sigue
-
Tipo A, mutación de ambos alelos de SLC3A1: Los heterocigotos muestran un patrón urinario de aminoácidos normal.
-
Tipo B, mutación de ambos alelos de SLC7A9: Los heterocigotos suelen mostrar un aumento de la excreción urinaria de cistina y aminoácidos dibásicos.
-
Tipo AB, cistinuria causada por 1 mutación en SLC3A1 y 1 mutación en SLC7A9: La cistinuria de tipo mixto puede estar causada por la interacción de 2 genes mutantes distintos, y la proteína codificada por el gen 19q interactúa directamente con rBAT en el segmento S3 del túbulo proximal (véase la Tabla).
Martens et al (2008) informaron recientemente de 3 síndromes de deleción génica asociados a la cistinuria tipo A: el síndrome de deleción 2p21, el síndrome de hipotonía-cistinuria (HCS) y una forma atípica del síndrome de hipotonía-cistinuria. Ambos alelos de SLC3A1 y PREPL faltan en los pacientes con HCS. Un gen adicional (C2orf34) está suprimido en el HCS atípico.
Tabla. Clasificación de la cistinuria (Abrir tabla en una ventana nueva)
|
Rosenberg et al |
Tipo I |
Tipo II |
Tipo III |
|
Molecular |
Tipo I |
No tipo I |
|
|
Gen responsable |
SLC3A1 |
SLC7A9 |
|
|
Banda |
2p21 |
19q13.1 |
|
|
Número de mutaciones |
>60 |
||
|
Mutación más común |
M467 |
V170M |
|
|
Población afectada |
Españoles mediterráneos, 40% |
Judíos libios |
|
|
Tasa de eliminación |
54% |
25% |
|
|
Proteína |
rBAT |
BAT1 |
|
|
Sistema de transporte de aminoácidos |
|||
|
Localización en el túbulo proximal convertido |
S3 |
S1, S2 |
|
|
Característica del transportador |
Alta afinidad, baja capacidad |
Baja afinidad, alta capacidad |
|
|
Características clínicas |
|||
|
Homocigotos |
Sintomáticos |
aproximadamente 90% sintomáticos |
|
|
Heterocigotos |
Sintomáticos |
aproximadamente 10%-13% sintomáticos |
|
|
Niveles de cistina en orina |
Normales |
Elevados +++++ |
Elevados + |
|
Niveles de cistina en plasma tras una prueba de carga oral |
Igual |
Igual o ligera subida |
Aumentada |
|
Transporte intestinal |
Absistente (no hay transporte de cistina, lisina, o arginina) |
Absistente |
Reducido |
Las pruebas recientes sugieren que el complejo 4F2HC/4F2LC explica para el sistema de transporte de aminoácidos Y+L en la superficie basolateral de las células tubulares proximales intestinales y renales y que las mutaciones del gen 4F2LC (SLC7A7) en la banda 14q11-13 causan la rara enfermedad recesiva llamada intolerancia a las proteínas de lisina.
Resumen
El rBAT, una glicoproteína de tipo II de 90 kd, es un transportador de alta afinidad e independiente del sodio para los aminoácidos dibásicos en los túbulos renales contorneados proximales de los roedores.El gen humano rBAT se ha localizado en la banda 2p21. Curiosamente, el análisis de vinculación sugiere que se trata de la misma región en la que se ha identificado un locus cistinúrico, el SLC3A1.Se han identificado 160 mutaciones diferentes en el gen SLC3A1 y 116 en el gen SLC7A9 en pacientes con cistinuria en todo el mundo.
La cistinuria tipo III y II (no tipo I) se ha relacionado con la banda 19p13.1 (SLC7A9); sin embargo, se necesitan más estudios para determinar el papel exacto del gen SLC7A9. Aproximadamente el 50% de los niños con 2 mutaciones de SLC3A1 (cistinuria clásica homocigota de tipo I) desarrollan al menos un cálculo en la primera década de vida.