A cisztin renális transzportja
A glomerulus könnyen szűri az aminosavakat, és a proximális tubuláris sejtek közel teljes reabszorpciót végeznek. A szűrt cisztinnek csupán 0,4%-a jelenik meg a vizeletben. Különböző szerzők tanulmányozták az aminosav-transzportot emberek, patkányok és nyulak proximális vesetubulusából nyert sejtmembránokban. Legalább 2 transzportrendszer felelős a cisztin reabszorpciójáért, a következők szerint:
-
Nagy affinitású rendszer: Ez a rendszer cisztinúriában szenvedő személyeknél érintett. A nagy affinitású rendszer közvetíti az L-cisztin és a dibázikus aminosavak 10%-ának felvételét a proximális tubulus egyenes harmadik szegmensének (S3) apikális membránjánál.
Kis affinitású rendszer: Ez a rendszer a proximális tubulus S1-S2 szakaszán van jelen, és az L-cisztin reabszorpció 90%-áért felelős. Az alacsony affinitású folyamat kiegészíti a magas affinitású folyamatot. A felszívódás után minden egyes cisztinmolekula intracellulárisan 2 molekula ciszteinné alakul át. A cisztein a bazolaterális membránon távozik.
A cisztinúriás családokból származó DNS genetikai vizsgálatai a 2. kromoszómán található hibás gént mutatják ki. A cisztin-transzportert kódoló gént, amelyet kezdetben rBAT-nak neveztek, a nemzetközi genom-adatbázisban ma SLC3A1 (SLC az oldottanyag-hordozót jelenti) néven ismerik. A 19. kromoszómán található második cisztinúria gén neve SLC7A9. A normál SLC7A9 gén a cisztin-transzporter b 0,+ AT (aminosav transzporter) nevű alegységét kódolja. A cisztinfelvétel folyamatát az SLC3A1 és az SLC7A9 gén termékei aktiválják. Az L-cisztin transzportja a kefe-határmembrán vezikulában nátrium-független és elektrogenikus. A cisztinuriás személyeknél a cisztin vagy a cisztein mozgása a tubuláris sejtekből a vérbe nem érintett.
A cisztin intesztinális transzportja
A nagy affinitású transzporter a jejunum apikális kefe-határmembránjában van jelen, és a cisztin és a kétbázisú aminosavak felszívódásáért felelős. A legtöbb cisztinuriás betegnél a cisztin felszívódása károsodott; a cisztinhiány azonban klinikailag nem jelentős, mivel a rövid láncú aminosavak felszívódását nem befolyásolja.
Normális esetben a cisztin és a többi dibázikus aminosav (pl. ornitin, lizin, arginin) a glomerulusban szűrődik és a proximális konvolutált tubulusban egy nagy affinitású luminális transzmembráncsatornán keresztül visszaszívódik. Ennek a csatornának a hibái a vizeletben a dibázikus aminosav szekréciójának emelkedett szintjét okozzák. Míg az ornitin, a lizin és az arginin teljesen oldható, a cisztin viszonylag oldhatatlan a vizelet 5-7-es fiziológiás pH-szintjén, 8,3-as pKa-szinttel. A vizelet 7,8-as és 8-as pH-szintjén a cisztin megfelelő oldhatósága majdnem megduplázódik, illetve megháromszorozódik.
Dent és Senior kimutatták, hogy a cisztin oldhatósága pH-függő. A cisztin oldhatósága a vizeletben körülbelül 250 mg/L (1 mmol/L) 7-es pH-szintig, de az oldhatóság magasabb pH-szinttel akár 500 mg/L (2 mmol/L) vagy annál is többre nő 7,5-es pH-szint felett, ahogyan az alábbi képen látható. A vizeletben végzett mérések egyértelműen kimutatták, hogy a cisztin oldhatósága lineárisan nő az ionerősség növekedésével. Pak és munkatársai kimutatták, hogy körülbelül 70 mg további cisztin oldható fel minden egyes liter oldatban az ionerősség 0,005-0,3 közötti növekedésével. Ezenkívül azonos ionerősség és pH mellett a cisztin oldhatósága a jelenlévő elektrolit adott típusától függően változik. Lásd az alábbi képet:
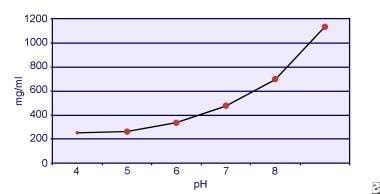 Cisztin oldhatósága a vizeletben.
Cisztin oldhatósága a vizeletben. Pak és Fuller 1983-ban végzett in vitro kísérletei kimutatták, hogy a legnagyobb oldékonyság kalcium-klorid jelenlétében érhető el, ezt követi a magnézium- és nátrium-klorid. Továbbá a cisztin oldhatóságát a vizelet makromolekulái is befolyásolják. Kimutatták, hogy a kolloid jelenléte a normál vizeletben növeli a cisztin oldhatóságát; e hatás mechanizmusa azonban nem világos. Mivel semmi sem gátolja a cisztin kristályosodását, a fő meghatározó tényező a vizelet szupertelítettsége. A kalcium-oxalát, a brushit vagy a hidroxiapatit heterogén nukleációja nem fordul elő cisztinuriás egyénekben.
A cisztinkristályosodás kockázati tényezői közé tartozik (1) az alacsony pH-szint, (2) a csökkent ionerősség, (3) a cisztinkristályok jelenléte és (4) a vizelet makromolekuláinak alacsony szintje.
A cisztein a cisztein diszulfidkötésű homodimerje és a következő szerkezettel rendelkezik:
COOH-CHNH2-CH2 -S-S-S-CH2-CHNH2- COOH Cisztein
COOH-CHNH2-CH2 -SH Cisztein
A cisztein a vékonybélben a veséhez hasonló módon szívódik fel. Cisztinuriás személyeknél a cisztin bélrendszeri felszívódása is különböző mértékben károsodott. A szérumcisztin másik forrása a metionin metabolizmusa. Két II-es típusú membránglikoprotein doménnek van szerepe az aminosav transzportban a plazmamembránon keresztül. Az első az rBAT, a második pedig a 4F2HC (a 4F2 antigén nehézlánca)
A cisztinuriás személyek kétharmadánál, akiknél kövek képződnek, tiszta cisztinkövek keletkeznek, egyharmadánál pedig cisztin- és kalcium-oxalátkő keveréke. Martins és munkatársai 2002-ben arról számoltak be, hogy a kalcium-oxalát kicsapódása nem a heterogén magképződés folyamatával, hanem egy másik anyagnak a rendszerbe történő hozzáadásával járó oldhatóságcsökkenéssel, azaz egy anyag kisózásával történik. A cisztinuriához gyakran társul hypocitraturia, hypercalciuria és hyperuricosuria is. Tekintettel viszonylag egységes, lamellás hasadási síkok nélküli kristályszerkezetükre, a tiszta cisztinkövek a Dretler-féle kő törékenységi indexen a legkeményebbek közé tartoznak.
A cisztinúria autoszomális-recesszív betegség, amely 3 altípusra osztható: Rosenberg I, II és III. Az I-es típusú cisztinúria a leggyakoribb változat, és a 2p16.3 sávra térképezték le. Az I-es típusú heterozigóták normális aminoaciduriát mutatnak. A klasszikus cisztinúria II. és III. típusát allélváltozatnak gondolták, de a kapcsolódási elemzések kimutatták, hogy a III. típus egy nem jellemzett gén (SLC7A9) hibája a 19q13.1 sávban. A II. és III. típus heterozigótái gyakran cisztinuriát manifesztálnak cisztinkövek nélkül, és fokozott kockázata lehet más típusú urolithiasisnak. Az I. típusú heterozigótákat a vizeletcisztin normális szintje különbözteti meg.
Az I. és II. típusú homozigótáktól eltérően a III. típusú homozigótáknál a plazma cisztinkoncentrációja orális cisztin beadása után megnő. Harris és munkatársai a cisztinúria genetikájának összetett természetéről számoltak be, amikor cisztinúria-probandák szüleinek (obligát heterozigóták) vizeletcisztin-kiválasztási szintjét mérték, és teljesen recesszív allélokat (mindkét szülő a referenciatartományban választ ki cisztint) és domináns allélokat (mindkét szülő magas szinten választ ki cisztint) találtak.
A cisztinúria klinikai osztályozásához a vizeletcisztin a probandus mindkét szülőjénél mérhető: I. fenotípus (recesszív, vizeletcisztinszint < 100 µmol/g kreatinin), II. fenotípus (domináns, vizeletcisztinszint >1000 µmol/g kreatinin) és III. fenotípus (részben domináns, vizeletcisztinszint 100-1000 µmol/g kreatinin). A cisztinúria a tünetek első megjelenésének életkora alapján is osztályozható (azaz csecsemőkori, fiatalkori, serdülőkori).
Egészséges egyéneknél a cisztin kiválasztás felső határa 20 mg/g kreatinin (< 10 µmol/mmol kreatinin). A homozigóták 400 mg/d-nál többet (1,7 mmol/d) választanak ki, és a homozigóta betegek cisztin-kiválasztása általában 600-1400 mg/d (2,5-5,8 mmol/d). Az I. és III. típusú cisztinúriában szenvedő heterozigóták 200 mg/d-nál kevesebbet (0,8 mmol/d) választanak ki, és nem képeznek köveket. A II. típusú heterozigóták akár 200-400 mg/d-t is kiválasztanak, de ezeknél a betegeknél kövek képződhetnek. A kőképződés gyakorisága nő, ha a vizelet cisztinkoncentrációja meghaladja a 700 µmol/l-t (170 mg/l).
Genetika
Az utóbbi években a molekuláris biológia fejlődésével új ismeretek gyűltek össze a cisztinúria patofiziológiájával kapcsolatban. 1992-ben több kutató beszámolt egy 2,3 kilobázisú renális cDNS (D2H vagy rBAT) expressziós klónozásáról, amely a cRNS-befecskendezett Xenopus laevis oocitákban a cisztin és a kétbázisú aminosavak nátrium-független felvételét indukálta. Az rBAT gént a 2. kromoszómára (2p21 sáv), a D2S119 és a D2S288 között térképeztük le. Ezt a gént a Genom Adatbázisban SLC3A1-nek nevezték el.
Immunohisztokémiai és in situ hibridizációs vizsgálatok kimutatták, hogy az rBAT a proximális tubulus és a vékonybél S3 (pars recta) szegmensének sejtjeiben a luminális ecsethatár membránjánál expresszálódik. 1995-ben Gasparini és munkatársai arról számoltak be, hogy az SLC3A1 mutációi I. típusú cisztinuriás betegeknél fordultak elő, II. vagy III. típusú cisztinuriás betegeknél nem. A mai napig több mint 160 különböző mutációt írtak le, beleértve a DNS-bázispárok kisebb és nagyobb delécióit is a génből. Az SLC3A1 egyik leggyakoribb genetikai elváltozását M467T-nek nevezik, és a legtöbb mutáció általában populáció-specifikus. Az M467T mutáció meglehetősen gyakori a mediterrán populációkban. Érdekes módon a mutációk 40%-át tette ki egy spanyol családi kohorszban, és ritka volt a kanadai Quebecben vizsgált betegeknél.
1999-ben izolálták az SLC7A9 (BAT1) gént. A gén egy 487 aminosavból álló fehérjét kódol, és a 19. kromoszómára (19q13 sáv) térképezték fel a D19S414 és a D19S220 között. Úgy tűnik, hogy a BAT1 termék egy membránfehérje, 12 membránon átívelő régióval. A BAT1 gén mutációi valószínűleg nem I. típusú cisztinúriát (Rosenberg II. és III. típus) okoznak. A 19q lokuszon lévő mutációk különösen gyakoriak a líbiai zsidók körében, és az urolithiasis kockázata azoknál a betegeknél, akik 2 ilyen 19q lokusz mutációt örökölnek, nagyjából hasonló, mint azoknál, akik 2 rBAT mutációt örökölnek.
116 mutációt jelentettek ebben a génben. A líbiai zsidóknál leggyakrabban előforduló mutáció azt eredményezte, hogy a fehérjében a valin aminosavmaradékot metionin váltotta fel (V170M). A V170M mutációval rendelkező heterozigótáknál a vizelet cisztinkoncentrációja 86-1238 µmol/g kreatinin között mozog. Így a V170M heterozigóták egyes értékei megfelelnek a III. típusú cisztinuriának, mások pedig a II. típusú cisztinuriának.
A II. és III. típusú cisztinuria közötti nyilvánvaló megkülönböztető jegy a II. típusú homozigótáknál a bélrendszeri cisztinfelszívódás hiánya. Pras 2000-ben molekuláris analízisen alapuló új osztályozást javasolt. A közelmúltban Dello Strologo és munkatársai új genetikai osztályozást javasoltak, az alábbiak szerint:
-
A típus, az SLC3A1 mindkét alléljának mutációja: A heterozigóták normális aminosav vizeletkiválasztást mutatnak.
-
B típus, az SLC7A9 mindkét alléljának mutációja: A heterozigóták általában fokozott cisztin- és dibázikus aminosav vizeletkiválasztást mutatnak.
-
AB típus, az SLC3A1 1 mutációja és az SLC7A9 1 mutációja által okozott cystinuria: A vegyes típusú cystinuriát 2 különböző mutáns gén kölcsönhatása okozhatja, és a 19q gén által kódolt fehérje közvetlenül kölcsönhatásba lép az rBAT-tal a proximális tubulus S3 szegmensében (lásd a táblázatot).
Martens és munkatársai (2008) nemrégiben 3 gén-deléciós szindrómáról számoltak be, amelyek az A típusú cisztinuriához társulnak: 2p21-deléciós szindróma, hipotónia-cisztinúria szindróma (HCS) és a hipotónia-cisztinúria szindróma atípusos formája. A HCS-ben szenvedő betegeknél az SLC3A1 és a PREPL mindkét allélja hiányzik. Az atipikus HCS-ben egy további gén (C2orf34) deletálódik.
Táblázat. A cisztinúria osztályozása (A táblázat megnyitása új ablakban)
Rosenberg et al
.
I. típus
II. típus
III. típus
Molekuláris
I. típus
Nem I. típus
felelős gén
.
SLC3A1
SLC7A9
Sáv
2p21
19q13.1
No. mutációk száma
>60
Leggyakoribb mutáció
.
M467
V170M
érintett népesség
mediterrán spanyol személyek, 40%
Líbiai zsidók
Deléciós arány
.
54%
25%
Protein
rBAT
BAT1
Aminosav transzport rendszer
Lokalizáció a proximális átalakult tubulusban
S3
S1, S2
Transzporter jellemző
magas affinitás, alacsony kapacitás
alacsony affinitás, nagy kapacitás
Klinikai jellemzők
Homozigóta
Tünetmentes
.
közelítőleg 90% tünetmentes
Heterozigóták
tünetmentes
közelítőleg 10%…13% tünetes
Húgyúti cisztinszint
Normális
Elemelkedett +++++
Elemelkedett +
Plazma cisztinszintje Orális terheléses vizsgálat után
Szintén
Szintén vagy enyhe emelkedés
Előre emelkedett
Bélrendszeri transzport
Nincs (nincs cisztin transzport, lizin, vagy arginin)
hiányzik
csökkent
Újabb bizonyítékok arra utalnak, hogy a 4F2HC/4F2LC komplex felelős a az Y+L aminosav transzport rendszerért felelős a bél és a vese proximalis tubuláris sejtjeinek basolaterális felszínén, és hogy a 4F2LC gén (SLC7A7) mutációi a 14q11- sávban található13 a lizin-fehérje intoleranciának nevezett ritka recesszív betegséget okozzák.
Összefoglaló
Az rBAT, egy 90 kd-os II-es típusú glikoprotein, egy nagy affinitású, nátrium-független transzporter a dibázikus aminosavak számára a proximális konvolúciós vese tubulusokban rágcsálókban. 2p21-es sávban lokalizálták a humán rBAT gént. Érdekes módon a kapcsolódási analízis arra utal, hogy ez ugyanaz a régió, ahol egy cisztinuriás lókuszt, az SLC3A1-t azonosították.Világszerte 160 különböző mutációt azonosítottak az SLC3A1 génben és 116-ot az SLC7A9 génben cisztinuriás betegeknél.
A III. és II. típusú cisztinúriát (nem I. típusú) a 19p13.1 sávhoz (SLC7A9) kapcsolták; az SLC7A9 gén pontos szerepének meghatározásához azonban további vizsgálatokra van szükség. A 2 SLC3A1 mutációval rendelkező gyermekek kb. 50%-ánál (klasszikus homozigóta I. típusú cystinuria) legalább egy kő alakul ki az élet első évtizedében.
 Cisztin oldhatósága a vizeletben.
Cisztin oldhatósága a vizeletben.