Trasporto renale della cistina
Gli aminoacidi sono facilmente filtrati dal glomerulo e subiscono un riassorbimento quasi completo dalle cellule tubulari prossimali. Solo lo 0,4% della cistina filtrata appare nelle urine. Diversi autori hanno studiato il trasporto degli amminoacidi nelle membrane cellulari ottenute dal tubulo renale prossimale di esseri umani, ratti e conigli. Almeno 2 sistemi di trasporto sono responsabili del riassorbimento della cistina, come segue:
-
Sistema ad alta affinità: Questo sistema è interessato nelle persone con cistinuria. Il sistema ad alta affinità media l’assorbimento del 10% di L-cistina e degli aminoacidi dibasici alla membrana apicale del terzo segmento rettilineo (S3) del tubulo prossimale.
-
Sistema a bassa affinità: Questo sistema è presente nella parte S1-S2 del tubulo prossimale ed è responsabile del 90% del riassorbimento della L-cistina. Il processo a bassa affinità aumenta il processo ad alta affinità. Dopo l’assorbimento, ogni molecola di cistina è convertita intracellularmente in 2 molecole di cisteina. La cisteina esce dalla membrana basolaterale.
Studi genetici sul DNA di famiglie con cistinuria rivelano un gene difettoso situato sul cromosoma 2. Il gene che codifica per il trasportatore di cistina, inizialmente chiamato rBAT, è ora conosciuto come SLC3A1 (SLC per trasportatore di soluti) nel database internazionale del genoma. Un secondo gene della cistinuria sul cromosoma 19 è chiamato SLC7A9. Il gene SLC7A9 normale codifica una subunità del trasportatore di cistina chiamata b 0,+ AT (trasportatore di aminoacidi). Il processo di assorbimento della cistina è attivato dai prodotti dei geni SLC3A1 e SLC7A9. Il trasporto di L-cistina nella vescicola della membrana del brush-border è indipendente dal sodio ed elettrogenico. Nelle persone con cistinuria, il movimento di cistina o cisteina dalle cellule tubulari al sangue non è influenzato.
Trasporto intestinale della cistina
Il trasportatore ad alta affinità è presente nella membrana a spazzola apicale del digiuno ed è responsabile dell’assorbimento della cistina e degli aminoacidi dibasici. La maggior parte dei pazienti con cistinuria ha un assorbimento alterato della cistina; tuttavia, la carenza di cistina non è clinicamente significativa perché l’assorbimento degli aminoacidi a catena corta non è interessato.
Normalmente, la cistina e gli altri aminoacidi dibasici (cioè, ornitina, lisina, arginina) sono filtrati al glomerulo e riassorbiti nel tubulo convoluto prossimale da un canale transmembrana luminale ad alta affinità. Difetti in questo canale causano elevati livelli di secrezione di aminoacidi dibasici nelle urine. Mentre l’ornitina, la lisina e l’arginina sono completamente solubili, la cistina è relativamente insolubile a livelli fisiologici di pH delle urine di 5-7, con un livello pKa di 8,3. A un livello di pH urinario di 7,8 e 8, la rispettiva solubilità della cistina è quasi raddoppiata e triplicata.
Dent e Senior hanno dimostrato che la solubilità della cistina dipende dal pH. La solubilità della cistina nelle urine è di circa 250 mg/L (1 mmol/L) fino a un livello di pH di 7, ma la solubilità aumenta con un livello di pH più alto fino a 500 mg/L (2 mmol/L) o più sopra un livello di pH di 7,5, come mostrato nell’immagine qui sotto. Le misurazioni nelle urine hanno chiaramente dimostrato che la solubilità della cistina aumenta linearmente con l’aumento della forza ionica. Pak e colleghi hanno dimostrato che circa 70 mg di cistina in più possono essere dissolti in ogni litro di soluzione, con un aumento della forza ionica da 0,005-0,3. Inoltre, a parità di forza ionica e di pH, la solubilità della cistina varia a seconda del particolare tipo di elettrolita presente. Vedere l’immagine qui sotto.
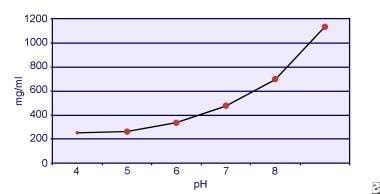 Solubilità della cistina nelle urine.
Solubilità della cistina nelle urine. Esperimenti in vitro di Pak e Fuller nel 1983 hanno rivelato che la più alta solubilità si ottiene in presenza di cloruro di calcio, seguito da cloruro di magnesio e di sodio. Inoltre, la solubilità della cistina è anche influenzata dalle macromolecole urinarie. È stato dimostrato che la presenza di colloidi nelle urine normali aumenta la solubilità della cistina; tuttavia, il meccanismo di questa azione non è chiaro. Poiché nulla inibisce la cristallizzazione della cistina, il principale determinante è la sovrasaturazione urinaria. La nucleazione eterogenea di ossalato di calcio, brushite o idrossiapatite non si verifica negli individui con cistinuria.
I fattori di rischio per la cristallizzazione della cistina includono (1) un basso livello di pH, (2) una ridotta forza ionica, (3) la presenza di cristalli di cistina e (4) bassi livelli di macromolecole urinarie.
La cistina è un omodimero della cisteina legato al disolfuro e ha la seguente struttura:
COOH-CHNH2-CH2 -S-S-CH2-CHNH2- COOH Cistina
COOH-CHNH2-CH2 -SH Cisteina
La cistina viene assorbita nell’intestino tenue in modo simile a quello dei reni. Nelle persone con cistinuria, l’assorbimento intestinale della cistina è anche alterato in vari gradi. Il metabolismo della metionina è un’altra fonte di cistina nel siero. Due domini di glicoproteine di membrana di tipo II sono stati implicati nel trasporto di aminoacidi attraverso la membrana plasmatica. Il primo è rBAT, e il secondo è 4F2HC (la catena pesante dell’antigene 4F2)
Due terzi delle persone con cistinuria che formano calcoli fanno calcoli di cistina pura, e un terzo hanno una miscela di calcoli di cistina e ossalato di calcio. Nel 2002, Martins et al hanno riferito che la precipitazione dell’ossalato di calcio avviene tramite un processo di salting-out, cioè la riduzione della solubilità di una sostanza dovuta all’aggiunta di un’altra sostanza al sistema, piuttosto che tramite il processo di nucleazione eterogenea. Anche l’ipocitraturia, l’ipercalciuria e l’iperuricosuria sono frequentemente associate alla cistinuria. Data la loro struttura cristallina relativamente uniforme senza piani di scissione lamellari, i calcoli di cistina pura sono tra i più duri sull’indice di fragilità delle pietre di Dretler.
La cistinuria è una malattia autosomica recessiva divisa in 3 sottotipi: Rosenberg I, II e III. La cistinuria di tipo I è la variante più comune ed è stata mappata sulla banda 2p16.3. Gli eterozigoti di tipo I mostrano un’aminoaciduria normale. La cistinuria classica, i tipi II e III, si pensava fossero varianti alleliche, ma le analisi di linkage hanno rivelato che il tipo III è un difetto di un gene non caratterizzato (SLC7A9) sulla banda 19q13.1. Gli eterozigoti di tipo II e III spesso manifestano cistinuria senza calcoli di cistina e possono essere a maggior rischio per altri tipi di urolitiasi. Gli eterozigoti di tipo I si distinguono per i livelli normali di cistina urinaria.
A differenza degli omozigoti di tipo I e II, gli omozigoti di tipo III mostrano un aumento della concentrazione di cistina nel plasma dopo la somministrazione orale di cistina. Harris et al hanno riportato la natura complessa della genetica della cistinuria misurando il livello di escrezione urinaria di cistina nei genitori (eterozigoti obbligati) dei probandi di cistinuria e hanno trovato alleli completamente recessivi (entrambi i genitori escretavano cistina nel range di riferimento) e alleli dominanti (entrambi i genitori escretavano cistina ad alti livelli).
Per classificare clinicamente la cistinuria, la cistina urinaria può essere misurata in ogni genitore di un probando come fenotipo I (recessivo, livello di cistina urinaria < 100 µmol/g di creatinina), fenotipo II (dominante, livello di cistina urinaria >1000 µmol/g di creatinina) e fenotipo III (parzialmente dominante, livello di cistina urinaria 100-1000 µmol/g di creatinina). La cistinuria può anche essere classificata in base all’età in cui compaiono i primi sintomi (cioè, infantile, giovanile, adolescenziale).
Negli individui sani, il limite superiore di escrezione della cistina è 20 mg/g di creatinina (< 10 µmol/mmol di creatinina). Gli omozigoti espellono più di 400 mg/d (1,7 mmol/d), e l’escrezione di cistina nei pazienti omozigoti è di solito 600-1400 mg/d (2,5-5,8 mmol/d). Gli eterozigoti con cistinuria di tipo I e III secernono meno di 200 mg/d (0,8 mmol/d) e non formano calcoli. Gli eterozigoti di tipo II espellono fino a 200-400 mg/d, ma questi pazienti possono formare calcoli. L’incidenza della formazione di calcoli aumenta quando la concentrazione di cistina urinaria supera i 700 µmol/L (170 mg/L).
Genetica
Negli ultimi anni, con i progressi della biologia molecolare, si sono accumulate nuove conoscenze sulla fisiopatologia della cistinuria. Nel 1992, diversi ricercatori hanno riportato la clonazione di espressione di un cDNA renale di 2,3 chilobasi (D2H o rBAT) che induceva l’assorbimento indipendente dal sodio della cistina e degli aminoacidi dibasici in ovociti di Xenopus laevis iniettati con cRNA. Il gene rBAT è stato mappato sul cromosoma 2 (banda 2p21) tra D2S119 e D2S288. Questo gene è ora chiamato SLC3A1 nel Genome Database.
Studi di immunoistochimica e di ibridazione in situ hanno rivelato che rBAT è espresso nelle cellule del segmento S3 (pars recta) del tubulo prossimale e dell’intestino tenue in corrispondenza della membrana del brush border. Nel 1995, Gasparini et al hanno riferito che mutazioni in SLC3A1 si sono verificate in pazienti con cistinuria di tipo I e non in pazienti con cistinuria di tipo II o III. Ad oggi, sono state descritte più di 160 diverse mutazioni, incluse piccole e grandi delezioni di coppie di basi del DNA dal gene. Una delle alterazioni genetiche più comuni in SLC3A1 è chiamata M467T, e la maggior parte delle mutazioni tende ad essere specifica della popolazione. La mutazione M467T è abbastanza comune nelle popolazioni mediterranee. È interessante notare che rappresentava il 40% delle mutazioni in una coorte spagnola di famiglie ed era rara nei pazienti studiati in Quebec, Canada.
Nel 1999, il gene SLC7A9 (BAT1) è stato isolato. Il gene codifica una proteina di 487 aminoacidi ed è stato mappato sul cromosoma 19 (banda 19q13) tra D19S414 e D19S220. Il prodotto BAT1 sembra essere una proteina di membrana con 12 regioni che attraversano la membrana. Le mutazioni nel gene BAT1 probabilmente causano cistinuria non di tipo I (Rosenberg tipo II e III). Le mutazioni al locus 19q sono particolarmente comuni tra gli ebrei libici, e il rischio di urolitiasi nei pazienti che ereditano 2 mutazioni di questo locus 19q è approssimativamente comparabile a quello dei pazienti che ereditano 2 mutazioni rBAT.
116 mutazioni in questo gene sono state riportate. La mutazione più comune negli ebrei libici è risultata in una metionina che sostituisce il residuo aminoacidico valina (V170M) nella proteina. Negli eterozigoti con la mutazione V170M, le concentrazioni di cistina urinaria variano da 86-1238 µmol/g di creatinina. Quindi, alcuni dei valori negli eterozigoti V170M sono coerenti con la cistinuria di tipo III e altri con la cistinuria di tipo II.
Un’apparente caratteristica distintiva tra la cistinuria di tipo II e di tipo III è la mancanza di assorbimento intestinale della cistina negli omozigoti di tipo II. Nel 2000, Pras ha proposto una nuova classificazione basata sull’analisi molecolare. Recentemente, Dello Strologo et al hanno proposto una nuova classificazione genetica, come segue:
-
Tipo A, mutazione di entrambi gli alleli di SLC3A1: gli eterozigoti mostrano un pattern urinario di aminoacidi normale.
-
Tipo B, mutazione di entrambi gli alleli di SLC7A9: gli eterozigoti mostrano solitamente un aumento dell’escrezione urinaria di cistina e aminoacidi dibasici.
-
Tipo AB, cistinuria causata da 1 mutazione in SLC3A1 e 1 mutazione in SLC7A9: la cistinuria di tipo misto può essere causata dall’interazione di 2 geni mutanti distinti, e la proteina codificata dal gene 19q interagisce direttamente con rBAT nel segmento S3 del tubulo prossimale (vedi tabella).
Martens et al (2008) hanno recentemente riportato 3 sindromi da delezione genica associate alla cistinuria di tipo A: sindrome da delezione 2p21, sindrome da ipotonia-cistinuria (HCS) e una forma atipica di sindrome da ipotonia-cistinuria. Entrambi gli alleli di SLC3A1 e PREPL mancano nei pazienti con HCS. Un ulteriore gene (C2orf34) è cancellato nella HCS atipica.
Tabella. Classificazione della cistinuria (Aprire la tabella in una nuova finestra)
|
Rosenberg et al |
Tipo I |
Tipo II |
Tipo III |
|
Molecolare |
Tipo I |
Non-tipo I |
|
|
Gene responsabile |
SLC3A1 |
SLC7A9 |
|
|
Banda |
2p21 |
19q13.1 |
|
|
No. di mutazioni |
>60 |
||
|
Mutazione più comune |
M467 |
V170M |
|
|
Popolazione colpita |
Spagnoli mediterranei, 40% |
Ebrei libici |
|
|
Tasso di eliminazione |
54% |
25% |
|
|
Proteina |
rBAT |
BAT1 |
|
|
Sistema di trasporto degli aminoacidi |
|||
|
Localizzazione nel tubulo prossimale convertito |
S3 |
S1, S2 |
|
|
Trasportatore caratteristico |
Alta affinità, bassa capacità |
Bassa affinità, alta capacità |
|
|
Caratteristiche cliniche |
|||
|
Omozigoti |
Sintomatici |
circa il 90% sintomatico |
|
|
Eterozigoti |
Asintomatico |
circa il 10%-13% sintomatico |
|
|
Livelli di cistina urinaria |
Normale |
elevato +++++ |
elevato + |
|
Livelli plasmatici di cistina dopo un test di carico orale |
Stesso |
Stesso o leggero aumento |
Aumentato |
|
Trasporto intestinale |
Assente (nessun trasporto di cistina, lisina, o arginina) |
Assente |
Ridotto |
Recenti prove suggeriscono che il complesso 4F2HC/4F2LC rappresenta del sistema di trasporto degli aminoacidi Y+L sulla superficie basale delle cellule tubulari prossimali intestinali e renali e che le mutazioni del gene 4F2LC (SLC7A7) sulla banda 14q11-13 causano la rara malattia recessiva chiamata intolleranza alla lisina-proteina.
Sommario
rBAT, una glicoproteina di tipo II di 90 kd, è un trasportatore ad alta affinità, indipendente dal sodio, per gli aminoacidi dibasici nei tubuli renali prossimali convoluti nei roditori.Il gene umano rBAT è stato localizzato sulla banda 2p21. È interessante notare che l’analisi di linkage suggerisce che questa è la stessa regione in cui è stato identificato un locus cistinurico, SLC3A1.160 diverse mutazioni nel gene SLC3A1 e 116 nel gene SLC7A9 sono state identificate in pazienti con cistinuria in tutto il mondo.
La cistinuria di tipo III e II (non di tipo I) è stata collegata alla banda 19p13.1 (SLC7A9); tuttavia, sono necessari ulteriori studi per determinare il ruolo esatto del gene SLC7A9. Circa il 50% dei bambini con 2 mutazioni SLC3A1 (cistinuria classica omozigote di tipo I) sviluppa almeno un calcolo entro la prima decade di vita.