Renaler Transport von Cystin
Aminosäuren werden vom Glomerulus leicht gefiltert und von den proximalen Tubuluszellen fast vollständig resorbiert. Nur 0,4 % des gefilterten Cystins gelangen in den Urin. Verschiedene Autoren haben den Aminosäuretransport in Zellmembranen aus dem proximalen Nierentubulus von Menschen, Ratten und Kaninchen untersucht. Mindestens 2 Transportsysteme sind für die Rückresorption von Cystin verantwortlich:
-
Hochaffinitätssystem: Dieses System ist bei Personen mit Cystinurie beeinträchtigt. Das hochaffine System vermittelt die Aufnahme von 10 % des L-Cystins und der dibasischen Aminosäuren an der apikalen Membran des geraden dritten Segments (S3) des proximalen Tubulus.
-
Niedrig-affines System: Dieses System befindet sich im S1-S2-Teil des proximalen Tubulus und ist für 90 % der L-Cystin-Rückresorption verantwortlich. Der Prozess der niedrigen Affinität ergänzt den Prozess der hohen Affinität. Nach der Resorption wird jedes Cystinmolekül intrazellulär in 2 Cysteinmoleküle umgewandelt. Das Cystein tritt an der basolateralen Membran aus.
Genetische Untersuchungen der DNA von Familien mit Cystinurie haben ein defektes Gen auf Chromosom 2 ergeben. Das Gen, das für den Cystin-Transporter kodiert und ursprünglich als rBAT bezeichnet wurde, ist in der internationalen Genom-Datenbank jetzt als SLC3A1 (SLC für solute carrier) bekannt. Ein zweites Cystinurie-Gen auf Chromosom 19 trägt die Bezeichnung SLC7A9. Das normale SLC7A9-Gen kodiert für eine Untereinheit des Cystin-Transporters namens b 0,+ AT (Aminosäuretransporter). Der Prozess der Cystinaufnahme wird durch die Genprodukte SLC3A1 und SLC7A9 aktiviert. Der Transport von L-Cystin in den Vesikeln der Bürstengrenzmembran ist natriumunabhängig und elektrogen. Bei Personen mit Cystinurie ist die Bewegung von Cystin oder Cystein aus den Tubuluszellen ins Blut nicht beeinträchtigt.
Intestinaler Transport von Cystin
Der hochaffine Transporter befindet sich in der apikalen Bürstengrenzmembran des Jejunums und ist für die Aufnahme von Cystin und dibasischen Aminosäuren verantwortlich. Die meisten Patienten mit Cystinurie haben eine gestörte Absorption von Cystin; ein Cystinmangel ist jedoch klinisch nicht signifikant, da die Absorption kurzkettiger Aminosäuren nicht beeinträchtigt wird.
Normalerweise werden Cystin und die anderen dibasischen Aminosäuren (d.h. Ornithin, Lysin, Arginin) im Glomerulus gefiltert und im proximalen Tubulus durch einen hochaffinen luminalen Transmembrankanal resorbiert. Defekte in diesem Kanal führen zu einer erhöhten Ausscheidung von dibasischen Aminosäuren im Urin. Während Ornithin, Lysin und Arginin vollständig löslich sind, ist Cystin bei physiologischen Urin-pH-Werten von 5-7 relativ unlöslich und hat einen pKa-Wert von 8,3. Bei einem Urin-pH-Wert von 7,8 und 8 verdoppelt bzw. verdreifacht sich die Löslichkeit von Cystin nahezu.
Dent und Senior zeigten, dass die Löslichkeit von Cystin pH-abhängig ist. Die Löslichkeit von Cystin im Urin beträgt bis zu einem pH-Wert von 7 etwa 250 mg/L (1 mmol/L), aber die Löslichkeit steigt mit einem höheren pH-Wert auf bis zu 500 mg/L (2 mmol/L) oder mehr über einem pH-Wert von 7,5, wie in der Abbildung unten dargestellt. Messungen im Urin haben eindeutig gezeigt, dass die Löslichkeit von Cystin mit zunehmender Ionenstärke linear ansteigt. Pak und Kollegen zeigten, dass in jedem Liter Lösung etwa 70 mg zusätzliches Cystin gelöst werden können, wenn die Ionenstärke von 0,005-0,3 ansteigt. Darüber hinaus variiert die Löslichkeit von Cystin bei gleicher Ionenstärke und gleichem pH-Wert je nach Art des vorhandenen Elektrolyten. Siehe die folgende Abbildung.
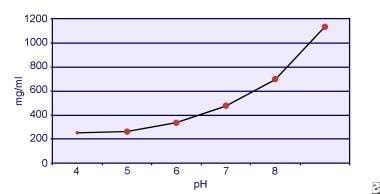 Cystinlöslichkeit im Urin.
Cystinlöslichkeit im Urin. In-vitro-Experimente von Pak und Fuller aus dem Jahr 1983 zeigten, dass die höchste Löslichkeit in Gegenwart von Calciumchlorid erreicht wird, gefolgt von Magnesium- und Natriumchlorid. Darüber hinaus wird die Löslichkeit von Cystin auch durch Makromoleküle im Urin beeinflusst. Das Vorhandensein von Kolloiden im normalen Urin erhöht nachweislich die Cystinlöslichkeit; der Mechanismus dieser Wirkung ist jedoch nicht klar. Da die Kristallisation von Cystin durch nichts gehemmt wird, ist der Hauptfaktor die Übersättigung des Urins. Heterogene Keimbildung von Calciumoxalat, Brushit oder Hydroxylapatit tritt bei Personen mit Cystinurie nicht auf.
Zu den Risikofaktoren für die Cystinkristallisation gehören (1) ein niedriger pH-Wert, (2) eine reduzierte Ionenstärke, (3) das Vorhandensein von Cystinkristallen und (4) geringe Mengen an Makromolekülen im Urin.
Cystin ist ein Disulfid-verknüpftes Homodimer von Cystein und hat folgende Struktur:
COOH-CHNH2-CH2 -S-S-CH2-CHNH2- COOH Cystin
COOH-CHNH2-CH2 -SH Cystein
Cystin wird im Dünndarm auf ähnliche Weise wie in den Nieren absorbiert. Bei Personen mit Cystinurie ist die intestinale Absorption von Cystin ebenfalls in unterschiedlichem Maße beeinträchtigt. Der Metabolismus von Methionin ist eine weitere Quelle für Serumcystin. Zwei Domänen von Typ-II-Membranglykoproteinen sind am Aminosäuretransport über die Plasmamembran beteiligt. Bei der ersten handelt es sich um rBAT, bei der zweiten um 4F2HC (die schwere Kette des 4F2-Antigens)
Zwei Drittel der Personen mit Cystinurie, die Steine bilden, haben reine Cystinsteine, und ein Drittel hat eine Mischung aus Cystin- und Calciumoxalatsteinen. Im Jahr 2002 berichteten Martins et al., dass die Ausfällung von Kalziumoxalat durch einen Aussalzungsprozess erfolgt, d. h. durch die Verringerung der Löslichkeit einer Substanz aufgrund der Zugabe einer anderen Substanz in das System, und nicht durch den Prozess der heterogenen Keimbildung. Hypocitraturie, Hypercalciurie und Hyperurikosurie sind ebenfalls häufig mit Cystinurie verbunden. Aufgrund ihrer relativ einheitlichen kristallinen Struktur ohne lamellierte Spaltflächen gehören reine Cystinsteine zu den härtesten auf dem Dretler’schen Steinbrüchigkeitsindex.
Die Cystinurie ist eine autosomal-rezessive Erkrankung, die in drei Subtypen unterteilt wird: Rosenberg I, II und III. Cystinurie Typ I ist die häufigste Variante und wurde auf Bande 2p16.3 kartiert. Heterozygote des Typs I weisen eine normale Aminosäureurie auf. Bei der klassischen Cystinurie, den Typen II und III, ging man davon aus, dass es sich um allelische Varianten handelt, aber Kopplungsanalysen haben ergeben, dass Typ III ein Defekt eines nicht charakterisierten Gens (SLC7A9) auf Bande 19q13.1 ist. Heterozygote der Typen II und III weisen häufig eine Zystinurie ohne Zystinsteine auf und können ein erhöhtes Risiko für andere Arten von Urolithiasis haben. Heterozygote des Typs I zeichnen sich durch normale Cystinwerte im Urin aus.
Im Gegensatz zu Homozygoten des Typs I und II zeigen Homozygote des Typs III nach oraler Verabreichung von Cystin einen Anstieg der Cystinkonzentration im Plasma. Harris et al. berichteten über die komplexe Natur der Genetik der Cystinurie, indem sie die Höhe der Cystinausscheidung im Urin bei den Eltern (obligate Heterozygoten) von Cystinurie-Probanden maßen und vollständig rezessive Allele (beide Elternteile scheiden Cystin im Referenzbereich aus) und dominante Allele (beide Elternteile scheiden Cystin in hohen Mengen aus) fanden.
Um die Cystinurie klinisch zu klassifizieren, kann Cystin im Urin bei jedem Elternteil eines Probanden als Phänotyp I (rezessiv, Cystin im Urin < 100 µmol/g Kreatinin), Phänotyp II (dominant, Cystin im Urin >1000 µmol/g Kreatinin) und Phänotyp III (teilweise dominant, Cystin im Urin 100-1000 µmol/g Kreatinin) gemessen werden. Die Cystinurie kann auch anhand des Alters, in dem die ersten Symptome auftreten, klassifiziert werden (d. h. im Kindes-, Jugend- oder Erwachsenenalter).
Bei gesunden Personen liegt die Obergrenze der Cystinausscheidung bei 20 mg/g Kreatinin (< 10 µmol/mmol Kreatinin). Homozygote scheiden mehr als 400 mg/d (1,7 mmol/d) aus, und die Cystinausscheidung bei homozygoten Patienten beträgt gewöhnlich 600-1400 mg/d (2,5-5,8 mmol/d). Heterozygote mit Cystinurie Typ I und III scheiden weniger als 200 mg/d (0,8 mmol/d) aus und bilden keine Steine. Heterozygote vom Typ II scheiden bis zu 200-400 mg/d aus, aber diese Patienten können Steine bilden. Die Häufigkeit der Steinbildung nimmt zu, wenn die Cystinkonzentration im Urin 700 µmol/L (170 mg/L) übersteigt.
Genetik
In den letzten Jahren haben sich mit den Fortschritten in der Molekularbiologie neue Erkenntnisse über die Pathophysiologie der Cystinurie ergeben. 1992 berichteten mehrere Forscher über die Expressionsklonierung einer 2,3-Kilobasen-Nieren-cDNA (D2H oder rBAT), die die natriumunabhängige Aufnahme von Cystin und dibasischen Aminosäuren in cRNA-injizierten Xenopus laevis-Oozyten induziert. Das rBAT-Gen wurde auf Chromosom 2 (Band 2p21) zwischen D2S119 und D2S288 kartiert. Dieses Gen wird in der Genomdatenbank nun als SLC3A1 bezeichnet.
Immunhistochemische und In-situ-Hybridisierungsstudien zeigten, dass rBAT in Zellen des S3-Segments (pars recta) des proximalen Tubulus und des Dünndarms an der luminalen Bürstengrenzmembran exprimiert wird. 1995 berichteten Gasparini et al., dass Mutationen in SLC3A1 bei Patienten mit Cystinurie Typ I und nicht bei Patienten mit Cystinurie Typ II oder III auftraten. Bis heute sind mehr als 160 verschiedene Mutationen beschrieben worden, darunter sowohl kleine als auch große Deletionen von DNA-Basenpaaren aus dem Gen. Eine der häufigsten genetischen Veränderungen in SLC3A1 heißt M467T, und die meisten Mutationen sind eher populationsabhängig. Die M467T-Mutation ist in mediterranen Populationen relativ häufig. Interessanterweise machte sie 40% der Mutationen in einer spanischen Familienkohorte aus und war bei Patienten, die in Quebec, Kanada, untersucht wurden, selten.
Im Jahr 1999 wurde das SLC7A9 (BAT1)-Gen isoliert. Das Gen kodiert ein 487-Aminosäuren-Protein und wurde auf Chromosom 19 (Band 19q13) zwischen D19S414 und D19S220 kartiert. Bei dem BAT1-Produkt scheint es sich um ein Membranprotein mit 12 membranüberspannenden Regionen zu handeln. Mutationen im BAT1-Gen verursachen wahrscheinlich eine Nicht-Typ-I-Cystinurie (Rosenberg-Typ II und III). Mutationen am 19q-Lokus sind unter libyschen Juden besonders häufig, und das Risiko einer Urolithiasis bei Patienten, die 2 solcher Mutationen am 19q-Lokus erben, ist in etwa vergleichbar mit dem von Patienten, die 2 rBAT-Mutationen erben.
116 Mutationen in diesem Gen wurden gemeldet. Die häufigste Mutation bei libyschen Juden führte dazu, dass ein Methionin den Valin-Aminosäurerest (V170M) im Protein ersetzte. Bei Heterozygoten mit der V170M-Mutation schwanken die Cystinkonzentrationen im Urin zwischen 86-1238 µmol/g Kreatinin. Somit entsprechen einige der Werte bei V170M-Heterozygoten der Typ-III-Cystinurie und andere der Typ-II-Cystinurie.
Ein offensichtliches Unterscheidungsmerkmal zwischen Typ-II- und Typ-III-Cystinurie ist die fehlende intestinale Cystinresorption bei Typ-II-Homozygoten. Im Jahr 2000 schlug Pras eine neue Klassifizierung auf der Grundlage der Molekularanalyse vor. Kürzlich haben Dello Strologo et al. eine neue genetische Klassifizierung vorgeschlagen, die wie folgt lautet
-
Typ A, Mutation beider Allele von SLC3A1: Heterozygote zeigen ein normales Aminosäure-Harnmuster.
-
Typ B, Mutation beider Allele von SLC7A9: Heterozygote zeigen in der Regel eine erhöhte Ausscheidung von Cystin und dibasischen Aminosäuren im Urin.
-
Typ AB, Cystinurie verursacht durch 1 Mutation in SLC3A1 und 1 Mutation in SLC7A9: Die Cystinurie vom Mischtyp kann durch das Zusammenwirken von 2 verschiedenen mutierten Genen verursacht werden, und das vom 19q-Gen kodierte Protein interagiert direkt mit rBAT im S3-Segment des proximalen Tubulus (siehe Tabelle).
Martens et al. (2008) berichteten kürzlich über drei Gendeletionssyndrome, die mit Cystinurie vom Typ A assoziiert sind: 2p21-Deletionssyndrom, Hypotonie-Cystinurie-Syndrom (HCS) und eine atypische Form des Hypotonie-Cystinurie-Syndroms. Bei Patienten mit HCS fehlen beide Allele von SLC3A1 und PREPL. Ein zusätzliches Gen (C2orf34) ist bei atypischem HCS deletiert.
Tabelle. Klassifikation der Cystinurie (Tabelle in einem neuen Fenster öffnen)
|
Rosenberg et al |
Typ I |
Typ II |
Typ III |
|
Molekular |
Typ I |
Nicht-Typ I |
|
|
Verantwortliches Gen |
SLC3A1 |
SLC7A9 |
|
|
Band |
2p21 |
19q13.1 |
|
|
Anzahl. der Mutationen |
>60 |
||
|
Häufigste Mutation |
M467 |
V170M |
|
|
Betroffene Bevölkerung |
Mittelmeerspanische Personen, 40% |
Libysche Juden |
|
|
Löschungsrate |
54% |
25% |
|
|
Protein |
rBAT |
BAT1 |
|
|
Aminosäure-Transportsystem |
|||
|
Lokalisierung im proximalen umgewandelten Tubulus |
S3 |
S1, S2 |
|
|
Transportereigenschaft |
Hohe Affinität, geringe Kapazität |
Niedrige Affinität, hohe Kapazität |
|
|
Klinische Merkmale |
|||
|
Homozygote |
Symptomatisch |
etwa 90% symptomatisch |
|
|
Heterozygote |
Asymptomatisch |
etwa 10%-13% symptomatisch |
|
|
Harn-Cystinspiegel |
Normal |
Erhöht +++++ |
Erhöht + |
|
Plasma-Cystinspiegel nach einem oralen Belastungstest |
Gleich |
Gleicher oder leichter Anstieg |
Erhöht |
|
Intestinaler Transport |
Absent (kein Transport von Cystin, Lysin, oder Arginin) |
Abwesend |
Reduziert |
Neue Erkenntnisse legen nahe, dass der 4F2HC/4F2LC-Komplex für das Y+L-Aminosäure-Transportsystem an der basolateralen Oberfläche der proximalen Tubuluszellen des Darms und der Nieren verantwortlich ist und dass Mutationen des 4F2LC-Gens (SLC7A7) auf Band 14q11-13 die seltene rezessive Krankheit namens Lysin-Protein-Intoleranz verursachen.
Zusammenfassung
rBAT, ein 90-kd-Glykoprotein vom Typ II, ist ein hochaffiner, natriumunabhängiger Transporter für dibasische Aminosäuren in den proximalen Nierentubuli von Nagetieren. rBAT wurde auf Bande 2p21 lokalisiert. Interessanterweise deutet eine Kopplungsanalyse darauf hin, dass dies dieselbe Region ist, in der ein Cystinurus-Lokus, SLC3A1, identifiziert wurde.160 verschiedene Mutationen im SLC3A1-Gen und 116 im SLC7A9-Gen wurden bei Patienten mit Cystinurie weltweit identifiziert.
Cystinurie Typ III und II (nicht Typ I) wurden mit der Bande 19p13.1 (SLC7A9) in Verbindung gebracht; es sind jedoch weitere Studien erforderlich, um die genaue Rolle des SLC7A9-Gens zu bestimmen. Etwa 50 % der Kinder mit 2 SLC3A1-Mutationen (klassische homozygote Typ-I-Cystinurie) entwickeln innerhalb des ersten Lebensjahrzehnts mindestens einen Stein.