Renal transport af cystin
Aminosyrer filtreres let af glomerulus og undergår næsten fuldstændig reabsorption af proximale tubulære celler. Kun 0,4 % af den filtrerede cystin optræder i urinen. Forskellige forfattere har undersøgt aminosyretransport i cellemembraner fra den proximale nyretubulus hos mennesker, rotter og kaniner. Mindst 2 transportsystemer er ansvarlige for cystinreabsorptionen, som følger:
-
Høj-affinitetssystem: Dette system er påvirket hos personer med cystinuria. Høj-affinitetssystemet medierer optagelsen af 10 % af L-cystin og de dibasiske aminosyrer ved den apikale membran i det lige tredje segment (S3) af den proximale tubulus.
Lav-affinitetssystem: Dette system er til stede i S1-S2-delen af den proximale tubulus og er ansvarlig for 90 % af L-cystin-reabsorptionen. Den lav-affinitetsproces forstærker den høj-affinitetsproces. Efter absorptionen omdannes hvert cystinmolekyle intracellulært til 2 cysteinmolekyler. Cystein udgår ved den basolaterale membran.
Genetiske undersøgelser af DNA fra familier med cystinuri afslører et defekt gen, der er placeret på kromosom 2. Genet, der koder for cystintransporteren, som oprindeligt blev betegnet rBAT, er nu kendt som SLC3A1 (SLC for solute carrier) i den internationale genomdatabase. Et andet cystinuriagen på kromosom 19 kaldes SLC7A9. Det normale SLC7A9-gen koder for en underenhed af cystintransporteren kaldet b 0,+ AT (aminosyretransportør). Processen med optagelsen af cystin aktiveres af SLC3A1- og SLC7A9-genprodukterne. Transport af L-cystin i børste-grænse membranvesiklen er natriumuafhængig og elektrogenetisk. Hos personer med cystinuri er bevægelsen af cystin eller cystein fra de tubulære celler ud i blodet ikke påvirket.
Intestinal transport af cystin
Den høj-affinitetstransporter er til stede i den apikale børste-grænsmembran i jejunum og er ansvarlig for absorptionen af cystin og dibasiske aminosyrer. De fleste patienter med cystinuri har nedsat absorption af cystin; cystinmangel er dog ikke klinisk signifikant, fordi absorptionen af kortkædede aminosyrer ikke påvirkes.
Normalt filtreres cystin og de andre dibasiske aminosyrer (dvs. ornithin, lysin, argininin) ved glomerulus og reabsorberes i den proximale konvoluterede tubulus af en luminal transmembrankanal med høj affinitet. Defekter i denne kanal forårsager forhøjede niveauer af sekretion af dibasiske aminosyrer i urinen. Mens ornithin, lysin og arginin er fuldstændig opløselige, er cystin relativt uopløseligt ved fysiologiske pH-niveauer i urinen på 5-7 med en pKa-værdi på 8,3. Ved et pH-niveau i urinen på 7,8 og 8 er den respektive opløselighed af cystin næsten fordoblet og tredoblet.
Dent og Senior påviste, at opløseligheden af cystin er pH-afhængig. Opløseligheden af cystin i urin er ca. 250 mg/L (1 mmol/L) op til et pH-niveau på 7, men opløseligheden stiger med et højere pH-niveau med op til 500 mg/L (2 mmol/L) eller mere over et pH-niveau på 7,5, som det fremgår af nedenstående billede. Målinger i urin har klart vist, at cystinopløseligheden stiger lineært med stigende ionstyrke. Pak og kolleger viste, at der kan opløses ca. 70 mg ekstra cystin i hver liter opløsning ved en stigning i ionstyrken fra 0,005-0,3. Desuden varierer cystinopløseligheden ved samme ionstyrke og pH-værdi afhængigt af den særlige type elektrolyt, der er til stede. Se billedet nedenfor.
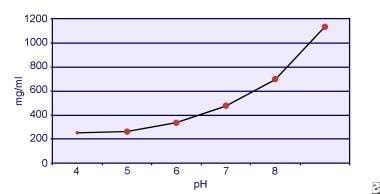 Cystinopløselighed i urin.
Cystinopløselighed i urin. In vitro-forsøg udført af Pak og Fuller i 1983 viste, at den højeste opløselighed opnås ved tilstedeværelse af calciumchlorid, efterfulgt af magnesium- og natriumchlorid. Endvidere påvirkes cystinopløseligheden også af makromolekyler i urinen. Tilstedeværelsen af kolloid i normal urin har vist sig at øge cystinopløseligheden; mekanismen bag denne virkning er imidlertid ikke klar. Da intet hæmmer cystinkrystallisationen, er den vigtigste determinant urinens overmætning. Heterogen kernedannelse af calciumoxalat, brushit eller hydroxyapatit forekommer ikke hos personer med cystinuri.
Risikofaktorer for cystinkrystallisation omfatter (1) lavt pH-niveau, (2) reduceret ionstyrke, (3) tilstedeværelse af cystinkrystaller og (4) lave niveauer af makromolekyler i urinen.
Cystin er en disulfidbundet homodimer af cystein og har følgende struktur:
COOH-CHNH2-CH2-CH2 -S-S-CH2-CHNH2-CHNH2- COOH Cystin
COOH-CHNH2-CH2-CH2 -SH Cystein
Cystin optages i tyndtarmen på en måde, der svarer til nyrernes. Hos personer med cystinuri er tarmabsorptionen af cystin også nedsat i varierende grad hos personer med cystinuri. Metabolisme af methionin er en anden kilde til serumcystin. To type II-membran-glykoproteindomæner er blevet inddraget i aminosyretransporten via plasmamembranen. Den første er rBAT, og den anden er 4F2HC (den tunge kæde af 4F2-antigenet)
To tredjedele af personer med cystinuri, der danner sten, laver rene cystinsten, og en tredjedel har en blanding af cystin- og calciumoxalatsten. I 2002 rapporterede Martins et al., at udfældning af calciumoxalat sker ved en saltningsproces, dvs. nedsættelse af et stofs opløselighed som følge af tilsætning af et andet stof til systemet, snarere end ved heterogen kernedannelsesproces. Hypocitraturi, hypercalciuri og hyperuricosuri er også ofte forbundet med cystinuri. På grund af deres relativt ensartede krystallinske struktur uden lamellerede spaltningsplaner er rene cystinkalk blandt de hårdeste på Dretlers stenfragilitetsindeks.
Cystinuria er en autosomal-recessiv sygdom, der er opdelt i 3 undertyper: Rosenberg I, II og III. Cystinuria type I er den mest almindelige variant og er blevet kortlagt til bånd 2p16.3. Type I heterozygoter viser normal aminoaciduri. Klassisk cystinuri, type II og III, blev anset for at være alleliske varianter, men koblingsanalyser har vist, at type III er en defekt i et ukarakteriseret gen (SLC7A9) på bånd 19q13.1. Heterozygoter af type II og III udviser ofte cystinuri uden cystinkalkuli og kan have en øget risiko for andre typer urolithiasis. Type I heterozygoter adskiller sig ved normale niveauer af urin-cystin.
I modsætning til type I og type II homozygoter viser type III homozygoter en stigning i plasmacystinkoncentrationen efter oral cystinadministration. Harris et al. rapporterede den komplekse karakter af genetikken af cystinuri ved at måle niveauet af urin-cystinudskillelse hos forældrene (obligatoriske heterozygoter) til cystinuri-probander og fandt fuldt recessive alleler (begge forældre udskiller cystin i referenceområdet) og dominerende alleler (begge forældre udskiller cystin på høje niveauer).
For at klassificere cystinuri klinisk kan urincystin måles hos hver forælder til en proband som fænotype I (recessiv, urincystinniveau < 100 µmol/g kreatinin), fænotype II (dominerende, urincystinniveau >1000 µmol/g kreatinin) og fænotype III (delvis dominerende, urincystinniveau 100-1000 µmol/g kreatinin). Cystinuri kan også klassificeres på grundlag af den alder, hvor symptomerne først optræder (dvs. infantil, juvenil, adolescent).
I raske personer er den øvre grænse for cystinudskillelse 20 mg/g kreatinin (< 10 µmol/mmol kreatinin). Homozygoter udskiller mere end 400 mg/d (1,7 mmol/d), og cystinudskillelsen hos homozygote patienter er normalt 600-1400 mg/d (2,5-5,8 mmol/d). Heterozygoter med type I- og III-cystinuri udskiller mindre end 200 mg/d (0,8 mmol/d) og danner ikke sten. Heterozygoter af type II udskiller op til 200-400 mg/d, men disse patienter kan danne sten. Forekomsten af stendannelse stiger, når cystinkoncentrationen i urinen overstiger 700 µmol/L (170 mg/L).
Genetik
I de seneste år er der med de fremskridt, der er sket inden for molekylærbiologi, kommet ny viden om cystinuriens patofysiologi. I 1992 rapporterede flere forskere om ekspressionsklonering af et 2,3-kilobase renalt cDNA (D2H eller rBAT), der inducerede natriumuafhængig optagelse af cystin og de dibasiske aminosyrer i cRNA-injicerede Xenopus laevis-ocytter. rBAT-genet blev kortlagt på kromosom 2 (bånd 2p21) mellem D2S119 og D2S288. Dette gen er nu navngivet SLC3A1 i Genome Database.
Immunohistokemiske og in situ hybridiseringsundersøgelser viste, at rBAT udtrykkes i celler i S3-segmentet (pars recta) i den proximale tubulus og tyndtarmen ved den luminale børste-grænsmembran. I 1995 rapporterede Gasparini et al., at mutationer i SLC3A1 forekom hos patienter med type I cystinuri og ikke hos patienter med type II eller III cystinuri. Til dato er der beskrevet mere end 160 forskellige mutationer, herunder både små og store deletioner af DNA-basepar fra genet. En af de mest almindelige genetiske ændringer i SLC3A1 kaldes M467T, og de fleste mutationer har en tendens til at være populationsspecifikke. M467T-mutationen er ret almindelig i middelhavsbefolkninger. Interessant nok udgjorde den 40 % af mutationerne i en spansk kohorte af familier og var sjælden hos patienter, der blev undersøgt i Quebec, Canada.
I 1999 blev SLC7A9 (BAT1)-genet isoleret. Genet koder for et protein med 487 aminosyrer og blev kortlagt på kromosom 19 (bånd 19q13) mellem D19S414 og D19S220. BAT1-produktet ser ud til at være et membranprotein med 12 membranoverskridende regioner. Mutationer i BAT1-genet forårsager sandsynligvis cystinuri uden for type I (Rosenberg type II og III). Mutationer på 19q-lokus er særligt almindelige blandt libyske jøder, og risikoen for urolithiasis hos patienter, der arver 2 sådanne 19q-lokusmutationer, er nogenlunde sammenlignelig med risikoen hos patienter, der arver 2 rBAT-mutationer.
116 mutationer i dette gen er blevet rapporteret. Den mest almindelige mutation hos libyske jøder resulterede i, at en methionin erstattede valin-aminosyrerest (V170M) i proteinet. Hos heterozygoter med V170M-mutationen varierer cystinkoncentrationerne i urinen fra 86-1238 µmol/g kreatinin. Således er nogle af værdierne hos V170M-heterozygoter i overensstemmelse med type III-cystinuri og andre med type II-cystinuri.
Et tilsyneladende kendetegn, der adskiller type II- og type III-cystinuri, er den manglende intestinale cystinabsorption hos type II-homozygoter. I 2000 foreslog Pras en ny klassifikation baseret på molekylær analyse. For nylig har Dello Strologo et al. foreslået en ny genetisk klassifikation som følger:
-
Type A, mutation af begge alleler af SLC3A1: Heterozygoter viser et normalt aminosyreurinmønster.
-
Type B, mutation af begge alleler af SLC7A9: Heterozygoter viser normalt en stigning i udskillelsen af cystin og dibasiske aminosyrer i urinen.
-
Type AB, cystinuri forårsaget af 1 mutation i SLC3A1 og 1 mutation i SLC7A9: Cystinuri af blandet type kan være forårsaget af interaktionen mellem 2 forskellige muterede gener, og det protein, der er kodet af 19q-genet, interagerer direkte med rBAT i S3-segmentet i den proximale tubulus (se tabellen).
Martens et al. (2008) rapporterede for nylig 3 gen-deletionssyndromer, der er forbundet med type A-cystinuri: 2p21-deletionssyndrom, hypotoni-cystinuriasyndrom (HCS) og en atypisk form af hypotoni-cystinuriasyndrom. Begge alleler af SLC3A1 og PREPL mangler hos patienter med HCS. Et yderligere gen (C2orf34) er slettet i atypisk HCS.
Tabel. Klassifikation af cystinuri (Åbn tabellen i et nyt vindue)
Rosenberg et al
Type I
Type II
Type III
Molekylær
Type I
Non-Type I
Responsibelt gen
SLC3A1
SLC7A9
Band
2p21
19q13.1
Nr. af mutationer
>60
Mest almindelige mutation
M467
V170M
Population, der er påvirket
Middelhavsspanske personer, 40%
Libiske jøder
Deletionsrate
54%
25%
Protein
rBAT
BAT1
Aminosyretransportsystem
Lokalisering i proximale konverterede tubuli
S3
S1, S2
Transportørkarakteristik
Høj affinitet, lav kapacitet
Lav affinitet, høj kapacitet
Kliniske karakteristika
Homozygoter
Symptomatisk
Omkring 90% symptomatisk
Heterozygoter
Asymptomatisk
Omkring 10%-13% symptomatiske
Urinære cystinniveauer
Normale
Hævede +++++
Hævede +
Plasma cystinniveauer efter en oral belastningstest
Samme
Samme eller let stigning
Stiget
Intestinaltransport
Absent (ingen transport af cystin, lysin, eller arginin)
Absent
Reduceret
Den seneste dokumentation tyder på, at 4F2HC/4F2LC-komplekset tegner sig for for Y+L-aminosyretransportsystemet på den basolaterale overflade af tarmens og nyrernes proximale tubulære celler, og at mutationer i 4F2LC-genet (SLC7A7) på bånd 14q11-13 forårsager den sjældne recessive sygdom kaldet lysin-proteinintolerance.
Resumé
rBAT, et 90-kd type II-glykoprotein, er en høj-affin, natrium-uafhængig transportør for dibasiske aminosyrer i de proximale konvolutterede nyretubuli hos gnavere. rBAT-genet hos mennesker er blevet lokaliseret på bånd 2p21. Interessant nok tyder en linkageanalyse på, at dette er den samme region, hvortil et cystinurisk locus, SLC3A1, er blevet identificeret.Der er blevet identificeret 160 forskellige mutationer i SLC3A1-genet og 116 i SLC7A9-genet hos patienter med cystinuri på verdensplan.
Type III og II cystinuri (ikke-type I) er blevet knyttet til bånd 19p13.1 (SLC7A9); der er dog behov for yderligere undersøgelser for at fastslå SLC7A9-genets nøjagtige rolle. Ca. 50 % af børn med 2 SLC3A1-mutationer (klassisk homozygot type I-cystinuri) udvikler mindst én sten inden for de første ti år af deres liv.
 Cystinopløselighed i urin.
Cystinopløselighed i urin.