Transport rénal de la cystine
Les acides aminés sont facilement filtrés par le glomérule et subissent une réabsorption quasi complète par les cellules tubulaires proximales. Seulement 0,4% de la cystine filtrée apparaît dans l’urine. Divers auteurs ont étudié le transport des acides aminés dans des membranes cellulaires obtenues à partir du tubule rénal proximal d’humains, de rats et de lapins. Au moins 2 systèmes de transport sont responsables de la réabsorption de la cystine, comme suit :
-
Système de haute affinité : Ce système est affecté chez les personnes atteintes de cystinurie. Le système à haute affinité assure la médiation de l’absorption de 10 % de la L-cystine et des acides aminés dibasiques au niveau de la membrane apicale du troisième segment droit (S3) du tubule proximal.
-
Système à faible affinité : Ce système est présent dans la partie S1-S2 du tubule proximal et est responsable de 90% de la réabsorption de la L-cystine. Le processus de basse affinité augmente le processus de haute affinité. Après absorption, chaque molécule de cystine est convertie au niveau intracellulaire en 2 molécules de cystéine. La cystéine sort au niveau de la membrane basolatérale.
Les études génétiques de l’ADN de familles atteintes de cystinurie révèlent un gène défectueux situé sur le chromosome 2. Le gène qui code pour le transporteur de la cystine, initialement appelé rBAT, est maintenant connu sous le nom de SLC3A1 (SLC pour solute carrier) dans la base de données internationale sur le génome. Un deuxième gène de la cystinurie sur le chromosome 19 est appelé SLC7A9. Le gène SLC7A9 normal code pour une sous-unité du transporteur de cystine appelée b 0,+ AT (transporteur d’acides aminés). Le processus d’absorption de la cystine est activé par les produits des gènes SLC3A1 et SLC7A9. Le transport de la L-cystine dans la vésicule de la membrane de la bordure en brosse est indépendant du sodium et électrogène. Chez les personnes atteintes de cystinurie, le déplacement de la cystine ou de la cystéine des cellules tubulaires vers le sang n’est pas affecté.
Transport intestinal de la cystine
Le transporteur à haute affinité est présent dans la membrane apicale de la bordure en brosse du jéjunum et est responsable de l’absorption de la cystine et des acides aminés dibasiques. La plupart des patients atteints de cystinurie présentent une altération de l’absorption de la cystine ; cependant, la déficience en cystine n’est pas cliniquement significative car l’absorption des acides aminés à chaîne courte n’est pas affectée.
Normalement, la cystine et les autres acides aminés dibasiques (c’est-à-dire l’ornithine, la de lysine, l’arginine) sont filtrés au niveau du glomérule et réabsorbés dans le tubule contourné proximal par un canal transmembranaire luminal de haute affinité. Les défauts de ce canal entraînent des niveaux élevés de sécrétion d’acides aminés dibasiques dans l’urine. Alors que l’ornithine, la lysine et l’arginine sont complètement solubles, la cystine est relativement insoluble à des niveaux de pH urinaire physiologique de 5 à 7, avec un pKa de 8,3. A un niveau de pH urinaire de 7,8 et 8, la solubilité respective de la cystine est presque doublée et triplée.
Dent et Senior ont démontré que la solubilité de la cystine est pH-dépendante. La solubilité de la cystine dans l’urine est d’environ 250 mg/L (1 mmol/L) jusqu’à un niveau de pH de 7, mais la solubilité augmente avec un niveau de pH plus élevé, jusqu’à 500 mg/L (2 mmol/L) ou plus au-dessus d’un niveau de pH de 7,5, comme le montre l’image ci-dessous. Les mesures effectuées dans l’urine ont clairement montré que la solubilité de la cystine augmente linéairement avec l’augmentation de la force ionique. Pak et ses collègues ont montré qu’environ 70 mg de cystine supplémentaire peuvent être dissous dans chaque litre de solution, avec une augmentation de la force ionique de 0,005-0,3. En outre, pour une même force ionique et un même pH, la solubilité de la cystine varie en fonction du type particulier d’électrolyte présent. Voir l’image ci-dessous.
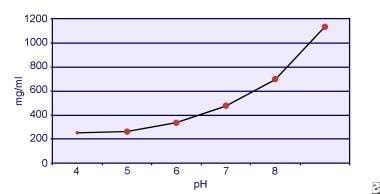 Solubilité de la cystine dans l’urine.
Solubilité de la cystine dans l’urine. Les expériences in vitro menées par Pak et Fuller en 1983 ont révélé que la solubilité la plus élevée est accomplie en présence de chlorure de calcium, suivi par le chlorure de magnésium et de sodium. En outre, la solubilité de la cystine est également affectée par les macromolécules urinaires. Il a été démontré que la présence de colloïdes dans l’urine normale augmente la solubilité de la cystine ; cependant, le mécanisme de cette action n’est pas clair. Comme rien n’inhibe la cristallisation de la cystine, le principal déterminant est la sursaturation urinaire. La nucléation hétérogène d’oxalate de calcium, de brushite ou d’hydroxyapatite ne se produit pas chez les personnes atteintes de cystinurie.
Les facteurs de risque de cristallisation de la cystine comprennent (1) un faible niveau de pH, (2) une force ionique réduite, (3) la présence de cristaux de cystine et (4) de faibles niveaux de macromolécules urinaires.
La cystine est un homodimère de la cystéine lié à un disulfure et présente la structure suivante :
COOH-CHNH2-CH2 -S-S-CH2-CHNH2- COOH Cystine
COOH-CHNH2-CH2 -SH Cystéine
La cystine est absorbée dans l’intestin grêle de manière similaire à celle des reins. Chez les personnes atteintes de cystinurie, l’absorption intestinale de la cystine est également altérée à des degrés divers. Le métabolisme de la méthionine est une autre source de cystine sérique. Deux domaines de glycoprotéines membranaires de type II ont été impliqués dans le transport des acides aminés via la membrane plasmique. Le premier est rBAT, et le second est 4F2HC (la chaîne lourde de l’antigène 4F2)
Les deux tiers des personnes atteintes de cystinurie qui forment des calculs font des calculs de cystine pure, et un tiers ont un mélange de calculs de cystine et d’oxalate de calcium. En 2002, Martins et al ont rapporté que la précipitation de l’oxalate de calcium se produit par un processus de salage, c’est-à-dire la réduction de la solubilité d’une substance due à l’ajout d’une autre substance dans le système, plutôt que par le processus de nucléation hétérogène. L’hypocitraturie, l’hypercalciurie et l’hyperuricosurie sont également fréquemment associées à la cystinurie. Compte tenu de leur structure cristalline relativement uniforme sans plans de clivage lamellés, les calculs de cystine pure sont parmi les plus durs selon l’indice de fragilité des calculs de Dretler.
La cystinurie est une maladie autosomique récessive divisée en 3 sous-types : Rosenberg I, II et III. La cystinurie de type I est la variante la plus courante et a été cartographiée sur la bande 2p16.3. Les hétérozygotes de type I présentent une aminoacidurie normale. On pensait que la cystinurie classique, les types II et III, étaient des variantes alléliques, mais les analyses de liaison ont révélé que le type III était une anomalie d’un gène non caractérisé (SLC7A9) sur la bande 19q13.1. Les hétérozygotes des types II et III manifestent souvent une cystinurie sans calcul de cystine et peuvent présenter un risque accru d’autres types d’urolithiase. Les hétérozygotes de type I se distinguent par des niveaux normaux de cystine urinaire.
Contrairement aux homozygotes de type I et II, les homozygotes de type III montrent une augmentation de la concentration de cystine dans le plasma après administration orale de cystine. Harris et al ont rapporté la nature complexe de la génétique de la cystinurie en mesurant le niveau d’excrétion de cystine urinaire chez les parents (hétérozygotes obligatoires) des probands cystinuriques et ont trouvé des allèles entièrement récessifs (les deux parents excrétaient de la cystine dans la plage de référence) et des allèles dominants (les deux parents excrétaient de la cystine à des niveaux élevés).
Pour classer cliniquement la cystinurie, la cystine urinaire peut être mesurée chez chaque parent d’un proband selon le phénotype I (récessif, taux de cystine urinaire < 100 µmol/g de créatinine), le phénotype II (dominant, taux de cystine urinaire >1000 µmol/g de créatinine) et le phénotype III (partiellement dominant, taux de cystine urinaire 100-1000 µmol/g de créatinine). La cystinurie peut également être classée en fonction de l’âge auquel les symptômes apparaissent pour la première fois (c’est-à-dire infantile, juvénile, adolescent).
Chez les individus sains, la limite supérieure d’excrétion de la cystine est de 20 mg/g de créatinine (<10 µmol/mmol de créatinine). Les homozygotes excrètent plus de 400 mg/j (1,7 mmol/j), et l’excrétion de cystine chez les patients homozygotes est généralement de 600-1400 mg/j (2,5-5,8 mmol/j). Les hétérozygotes atteints de cystinurie de type I et III excrètent moins de 200 mg/j (0,8 mmol/j) et ne forment pas de calculs. Les hétérozygotes de type II excrètent jusqu’à 200-400 mg/j, mais ces patients peuvent former des calculs. L’incidence de la formation de calculs augmente lorsque la concentration de cystine urinaire dépasse 700 µmol/L (170 mg/L).
Génétique
Ces dernières années, avec les progrès de la biologie moléculaire, de nouvelles connaissances se sont accumulées concernant la physiopathologie de la cystinurie. En 1992, plusieurs chercheurs ont rapporté le clonage d’expression d’un ADNc rénal de 2,3 kilobases (D2H ou rBAT) qui induisait une absorption indépendante du sodium de la cystine et des acides aminés dibasiques dans des ovocytes de Xenopus laevis injectés par l’ADNc. Le gène rBAT a été cartographié sur le chromosome 2 (bande 2p21) entre D2S119 et D2S288. Ce gène est maintenant nommé SLC3A1 dans la base de données du génome.
Des études immunohistochimiques et d’hybridation in situ ont révélé que rBAT est exprimé dans les cellules du segment S3 (pars recta) du tubule proximal et de l’intestin grêle au niveau de la membrane luminale de la bordure en brosse. En 1995, Gasparini et al ont signalé que des mutations de SLC3A1 se produisaient chez les patients atteints de cystinurie de type I et non chez les patients atteints de cystinurie de type II ou III. À ce jour, plus de 160 mutations différentes ont été décrites, y compris de petites et grandes délétions de paires de bases d’ADN du gène. L’une des altérations génétiques les plus courantes de SLC3A1 est appelée M467T, et la plupart des mutations ont tendance à être spécifiques à une population. La mutation M467T est assez courante dans les populations méditerranéennes. Fait intéressant, elle représentait 40 % des mutations dans une cohorte de familles espagnoles et était rare chez les patients étudiés au Québec, au Canada.
En 1999, le gène SLC7A9 (BAT1) a été isolé. Le gène code pour une protéine de 487 acides aminés et a été cartographié sur le chromosome 19 (bande 19q13) entre D19S414 et D19S220. Le produit BAT1 semble être une protéine membranaire comportant 12 régions membranaires. Les mutations du gène BAT1 sont probablement à l’origine de la cystinurie de type I (type II et III de Rosenberg). Les mutations au locus 19q sont particulièrement fréquentes chez les Juifs libyens, et le risque d’urolithiase chez les patients qui héritent de 2 de ces mutations au locus 19q est à peu près comparable à celui des patients qui héritent de 2 mutations rBAT.
116 mutations dans ce gène ont été signalées. La mutation la plus fréquente chez les Juifs libyens a entraîné le remplacement par une méthionine du résidu d’acide aminé valine (V170M) dans la protéine. Chez les hétérozygotes présentant la mutation V170M, les concentrations de cystine urinaire varient de 86 à 1 238 µmol/g de créatinine. Ainsi, certaines des valeurs chez les hétérozygotes V170M correspondent à une cystinurie de type III et d’autres à une cystinurie de type II.
Un trait distinctif apparent entre la cystinurie de type II et celle de type III est l’absence d’absorption intestinale de la cystine chez les homozygotes de type II. En 2000, Pras a proposé une nouvelle classification basée sur l’analyse moléculaire. Récemment, Dello Strologo et al ont proposé une nouvelle classification génétique, comme suit :
-
Type A, mutation des deux allèles de SLC3A1 : les hétérozygotes présentent un profil urinaire normal en acides aminés.
-
Type B, mutation des deux allèles de SLC7A9 : les hétérozygotes présentent généralement une augmentation de l’excrétion urinaire de cystine et d’acides aminés dibasiques.
-
Type AB, cystinurie causée par 1 mutation dans SLC3A1 et 1 mutation dans SLC7A9 : La cystinurie de type mixte peut être causée par l’interaction de 2 gènes mutants distincts, et la protéine codée par le gène 19q interagit directement avec rBAT dans le segment S3 du tubule proximal (voir le tableau).
Martens et al (2008) ont récemment signalé 3 syndromes de délétion génique associés à la cystinurie de type A : le syndrome de délétion 2p21, le syndrome d’hypotonie-cystinurie (HCS) et une forme atypique du syndrome d’hypotonie-cystinurie. Les deux allèles de SLC3A1 et PREPL sont absents chez les patients atteints du syndrome HCS. Un gène supplémentaire (C2orf34) est supprimé dans le HCS atypique.
Tableau. Classification de la cystinurie (Ouvrir le tableau dans une nouvelle fenêtre)
|
Rosenberg et al |
Type I |
Type II |
Type III |
|
Moléculaire |
Type I |
Non-Type I |
|
|
Gène responsable |
SLC3A1 |
SLC7A9 |
|
|
Bande |
2p21 |
19q13.1 |
|
|
No. de mutations |
>60 |
||
|
Mutation la plus fréquente |
M467 |
V170M |
|
|
Population touchée |
Personnes espagnoles méditerranéennes, 40% |
Juifs libyens |
|
|
Taux de délétion |
54% |
25% |
|
|
Protéine |
rBAT |
BAT1 |
|
|
Système de transport des acides aminés |
|||
|
Localisation dans le tubule converti proximal |
S3 |
S1, S2 |
|
|
Caractéristiques du transporteur |
Haute affinité, faible capacité |
Faible affinité, haute capacité |
|
|
Caractéristiques cliniques |
|||
|
Homozygotes |
Symptomatiques |
environ 90% de symptômes |
|
|
Hétérozygotes |
Asymptomatiques |
environ 10%-13% symptomatiques |
|
|
Cystinémie urinaire |
Normale |
Élevée +++++ |
Élevée + |
|
Cystinémie plasmatique après un test de charge oral |
Même |
Même ou légère augmentation |
Augmentation |
|
Transport intestinal |
Absent (pas de transport de cystine, lysine, ou d’arginine) |
Absent |
Réduit |
Des preuves récentes suggèrent que le complexe 4F2HC/4F2LC rend compte pour le système de transport des acides aminés Y+L à la surface basolatérale des cellules tubulaires proximales intestinales et rénales et que les mutations du gène 4F2LC (SLC7A7) sur la bande 14q11-.13 provoquent la maladie récessive rare appelée intolérance aux protéines de la lysine.
Résumé
La rBAT, une glycoprotéine de type II de 90 kd, est un transporteur de haute affinité, indépendant du sodium, pour les acides aminés dibasiques dans les tubules rénaux contournés proximaux des rongeurs.Le gène humain de la rBAT a été localisé sur la bande 2p21. Il est intéressant de noter que l’analyse de liaison suggère qu’il s’agit de la même région à laquelle un locus cystinurique, SLC3A1, a été identifié.160 mutations différentes dans le gène SLC3A1 et 116 dans le gène SLC7A9 ont été identifiées chez des patients atteints de cystinurie dans le monde entier.
La cystinurie de type III et II (non de type I) a été liée à la bande 19p13.1 (SLC7A9) ; toutefois, des études supplémentaires sont nécessaires pour déterminer le rôle exact du gène SLC7A9. Environ 50 % des enfants présentant 2 mutations de SLC3A1 (cystinurie classique homozygote de type I) développent au moins un calcul dans la première décennie de leur vie.