Transporte renal de cistina
Aminoácidos são prontamente filtrados pelo glomérulo e sofrem reabsorção quase completa pelas células tubulares proximais. Apenas 0,4% da cistina filtrada aparece na urina. Vários autores estudaram o transporte de aminoácidos nas membranas celulares obtidas do túbulo proximal renal de humanos, ratos e coelhos. Pelo menos 2 sistemas de transporte são responsáveis pela reabsorção da cistina, como segue:
-
Sistema de alta afinidade: Este sistema é afectado em pessoas com cistinúria. O sistema de alta afinidade medeia a absorção de 10% de L-cistina e dos aminoácidos dibásicos na membrana apical do terceiro segmento reto (S3) do túbulo proximal.
-
Sistema de baixa afinidade: Este sistema está presente na parte S1-S2 do túbulo proximal e é responsável por 90% da reabsorção de L-cystine. O processo de baixa afinidade aumenta o processo de alta afinidade. Após a absorção, cada molécula de cistina é convertida intracelularmente em 2 moléculas de cisteína. A cisteína sai na membrana basolateral.
Estudos genéticos de DNA de famílias com cistinúria revelam um gene defeituoso localizado no cromossoma 2. O gene que codifica o transportador de cistina, inicialmente denominado rBAT, é agora conhecido como SLC3A1 (SLC para transportador de soluto) no Banco de Dados de Genoma internacional. Um segundo gene de cistinúria no cromossomo 19 é chamado SLC7A9. O gene SLC7A9 normal codifica uma subunidade do transportador de cistina chamada b 0,+ AT (transportador de aminoácidos). O processo de captação da cistina é ativado pelos produtos dos genes SLC3A1 e SLC7A9. O transporte da cistina L na vesícula da membrana da borda da escova é independente do sódio e eletrogênico. Em pessoas com cistinúria, o movimento da cistina ou cisteína das células tubulares para o sangue não é afectado.
Transporte intestinal de cistina
O transportador de alta afinidade está presente na membrana apical do jejuno e é responsável pela absorção de cistina e aminoácidos dibásicos. A maioria dos pacientes com cistinúria tem dificuldade de absorção da cistina; entretanto, a deficiência de cistina não é clinicamente significativa porque a absorção de aminoácidos de cadeia curta não é afetada.
Normalmente, a cistina e os outros aminoácidos dibásicos (ou seja, ornitina, lisina, arginina) são filtrados no glomérulo e reabsorvidos no túbulo convoluto proximal por um canal transmembrana luminal de alta afinidade. Os defeitos neste canal causam níveis elevados de secreção de aminoácidos dibásicos na urina. Enquanto a ornitina, lisina e arginina são completamente solúveis, a cistina é relativamente insolúvel a níveis fisiológicos de pH da urina de 5-7, com um nível de pKa de 8,3. A um nível de pH da urina de 7,8 e 8, a respectiva solubilidade da cistina é quase dobrada e triplicada.
Dent e Senior demonstraram que a solubilidade da cistina é dependente do pH. A solubilidade da cistina na urina é aproximadamente 250 mg/L (1 mmol/L) até um nível de pH de 7, mas a solubilidade aumenta com um nível de pH mais alto em até 500 mg/L (2 mmol/L) ou mais acima de um nível de pH de 7,5, como mostrado na imagem abaixo. As medições na urina mostraram claramente que a solubilidade da cistina aumenta linearmente com o aumento da força iónica. Pak e colegas mostraram que aproximadamente 70 mg de cistina adicional podem ser dissolvidos em cada litro de solução, com um aumento na força iônica de 0,005-0,3. Além disso, com a mesma força iônica e pH, a solubilidade da cistina varia de acordo com o tipo particular de eletrólito presente. Veja a imagem abaixo.
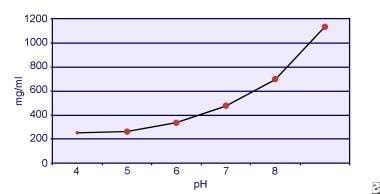 Solubilidade da cistina na urina.
Solubilidade da cistina na urina. Experiências in vitro de Pak e Fuller em 1983 revelaram que a maior solubilidade é conseguida na presença de cloreto de cálcio, seguido pelo cloreto de magnésio e sódio. Além disso, a solubilidade da cistina é também afetada pelas macromoléculas urinárias. A presença de colóide na urina normal tem demonstrado aumentar a solubilidade da cistina; no entanto, o mecanismo desta acção não é claro. Como nada inibe a cristalização da cistina, o principal determinante é a supersaturação urinária. A nucleação heterogênea de oxalato de cálcio, brushite ou hidroxiapatita não ocorre em indivíduos com cistinúria.
Os fatores de risco para cristalização de cistina incluem (1) baixo nível de pH, (2) força iônica reduzida, (3) presença de cristais de cistina, e (4) baixos níveis de macromoléculas urinárias.
Cistina é um homodímero dissulfidado de cisteína e tem a seguinte estrutura:
COOH-CHNH2-CH2 -S-S-CH2-CHNH2- COOH Cystine
COOH-CHNH2-CH2 -SH Cisteína
Cistina é absorvida no intestino delgado de forma semelhante à dos rins. Em pessoas com cistinúria, a absorção intestinal da cistina também é prejudicada em vários graus. O metabolismo da metionina é outra fonte de cistina sérica. Dois domínios de glicoproteínas da membrana tipo II têm sido implicados no transporte de aminoácidos através da membrana plasmática. O primeiro é rBAT, e o segundo é 4F2HC (a cadeia pesada do antígeno 4F2)
Dois terços das pessoas com cistinúria que formam pedras fazem cálculos de cistina pura, e um terço tem uma mistura de cálculos de cistina e oxalato de cálcio. Em 2002, Martins et al reportaram que a precipitação de oxalato de cálcio ocorre por um processo de salga, ou seja, a redução da solubilidade de uma substância devido à adição de outra substância ao sistema, e não pelo processo de nucleação heterogênea. Hipocitratúria, hipercalciúria e hiperuricosúria também são frequentemente associadas à cistinúria. Dada a sua estrutura cristalina relativamente uniforme sem planos de clivagem lamelada, os cálculos de cistina pura estão entre os mais difíceis no índice de fragilidade da pedra de Dretler.
Cistinúria é uma doença autossômica recessiva dividida em 3 subtipos: Rosenberg I, II, e III. A cistinúria tipo I é a variante mais comum e foi mapeada para a banda 2p16.3. Os heterozigotos do tipo I mostram aminoaciduria normal. Cistinúria clássica, tipos II e III, foram consideradas variantes alélicas, mas análises de ligação revelaram que o tipo III é um defeito de um gene não caracterizado (SLC7A9) na banda 19q13.1. Heterozigotos dos tipos II e III frequentemente manifestam cistinúria sem cálculos cistinosos e podem estar em risco aumentado para outros tipos de urolitíase. Os heterozigotos tipo I são distinguidos pelos níveis normais de cistina urinária.
Os homozigotos tipo I e tipo II não semelhantes, os homozigotos tipo III mostram um aumento na concentração plasmática de cistina após a administração oral de cistina. Harris et al. relataram a natureza complexa da genética da cistinúria medindo o nível de excreção de cistina urinária nos progenitores (heterozigotos obrigatórios) dos probandos de cistinúria e encontraram alelos totalmente recessivos (ambos os progenitores excretaram cistina na faixa de referência) e alelos dominantes (ambos os progenitores excretaram cistina em níveis elevados).
Para classificar clinicamente a cistina, a cistina urinária pode ser medida em cada um dos pais de uma probanda como fenótipo I (recessivo, nível de cistina urinária < 100 µmol/g de creatinina), fenótipo II (dominante, nível de cistina urinária >1000 µmol/g de creatinina), e fenótipo III (parcialmente dominante, nível de cistina urinária 100-1000 µmol/g de creatinina). A cistinúria também pode ser classificada com base na idade em que os sintomas aparecem pela primeira vez (ou seja, infantil, juvenil, adolescente).
Em indivíduos saudáveis, o limite superior para excreção de cistina é de 20 mg/g de creatinina (< 10 µmol/mmol de creatinina). Homozigotos excretam mais de 400 mg/d (1,7 mmol/d), e a excreção de cistina em pacientes homozigotos é geralmente 600-1400 mg/d (2,5-5,8 mmol/d). Heterozigotos com cistinúria tipo I e III excretam menos de 200 mg/d (0,8 mmol/d) e não formam pedras. Os heterozigotos do tipo II excretam até 200-400 mg/d, mas estes pacientes podem formar pedras. A incidência da formação de cálculos aumenta quando a concentração de cistina urinária excede 700 µmol/L (170 mg/L).
Genetics
Nos últimos anos, com os avanços na biologia molecular, novos conhecimentos se acumularam com relação à fisiopatologia da cistinúria. Em 1992, vários investigadores relataram a clonagem de expressão de um cDNA renal de 2,3-kilobase (D2H ou rBAT) que induziu a absorção independente de sódio da cistina e dos aminoácidos dibásicos nos oócitos Xenopus laevis injetados por cRNA. O gene rBAT foi mapeado para o cromossomo 2 (banda 2p21) entre D2S119 e D2S288. Este gene é agora denominado SLC3A1 no Genome Database.
Immunohistoquímica e estudos de hibridação in situ revelaram que o rBAT é expresso em células do segmento S3 (pars recta) do túbulo proximal e intestino delgado na membrana da borda da escova luminal. Em 1995, Gasparini et al. relataram que mutações no SLC3A1 ocorreram em pacientes com cistinúria tipo I e não em pacientes com cistinúria tipo II ou III. Até o momento, mais de 160 mutações diferentes foram descritas, incluindo pequenas e grandes deleções de pares de base de DNA do gene. Uma das alterações genéticas mais comuns no SLC3A1 é chamada M467T, e a maioria das mutações tende a ser específica da população. A mutação M467T é bastante comum em populações mediterrâneas. Curiosamente, ela foi responsável por 40% das mutações em uma coorte espanhola de famílias e foi rara em pacientes estudados no Quebec, Canadá.
Em 1999, o gene SLC7A9 (BAT1) foi isolado. O gene codifica uma proteína 487-aminoácida e foi mapeado para o cromossomo 19 (banda 19q13) entre D19S414 e D19S220. O produto BAT1 parece ser uma proteína de membrana com 12 regiões de membranas. Mutações no gene BAT1 provavelmente causam cistinúria não do tipo I (Rosenberg tipo II e III). Mutações no locus 19q são especialmente comuns entre os judeus líbios, e o risco de urolitíase em pacientes que herdam 2 mutações no locus 19q é aproximadamente comparável ao de pacientes que herdam 2 mutações rBAT.
116 mutações neste gene têm sido relatadas. A mutação mais comum nos judeus líbios resultou em uma metionina substituindo o resíduo do aminoácido valina (V170M) na proteína. Em heterozigotos com a mutação V170M, as concentrações de cistina urinária variam de 86-1238 µmol/g de creatinina. Assim, alguns dos valores em heterozigotos V170M são consistentes com cistinúria tipo III e outros com cistinúria tipo II.
Uma característica distintiva aparente entre cistinúria tipo II e tipo III é a falta de absorção intestinal de cistina em homozigotos tipo II. Em 2000, Pras sugeriu uma nova classificação baseada na análise molecular. Recentemente, Dello Strologo et al propuseram uma nova classificação genética, como se segue:
-
Tipo A, mutação de ambos os alelos de SLC3A1: Heterozigotos mostram um padrão urinário normal de aminoácidos.
Tipo B, mutação de ambos os alelos de SLC7A9: Heterozigotos normalmente mostram um aumento da excreção urinária de cistina e aminoácidos dibásicos.
Type AB, cistinúria causada por 1 mutação em SLC3A1 e 1 mutação em SLC7A9: Cistinúria do tipo misto pode ser causada pela interação de 2 genes mutantes distintos, e a proteína codificada pelo gene 19q interage diretamente com o rBAT no segmento S3 do túbulo proximal (veja a Tabela).
Martens et al (2008) reportaram recentemente 3 síndromes de eliminação de genes associados com cistinúria tipo A: síndrome de eliminação 2p21, síndrome de hipotonia-cistinúria (HCS), e uma forma atípica de síndrome de hipotonia-cistinúria. Ambos os alelos de SLC3A1 e PREPL estão ausentes em pacientes com HCS. Um gene adicional (C2orf34) é deletado na HCS atípica.
Tabela. Classificação da Cistinúria (Abrir tabela em uma nova janela)
Rosenberg et al
Tipo I
Tipo II
Tipo III
Molecular
Tipo I
Não-Tipo I
Gene responsável
SLC3A1
SLC7A9
Band
2p21
19q13.1
Não. de mutações
>60
Mutação mais comum
M467
V170M
População afetada
Espanholes mediterrâneos, 40%
Judeus da Líbia
> Taxa de depleção
54%
25%
Proteína
rBAT
BAT1
Sistema de transporte de aminoácidos
Localização em túbulo proximal convertido
S3
S1, S2
Característica do transportador
Alta afinidade, baixa capacidade
Baixa afinidade, alta capacidade
Características clínicas
Homozygotes
Sintomático
aproximadamente 90% sintomático
Heterozigotos
Asintomático
aproximadamente 10%-13% sintomático
Níveis de cistina urinária
Normal
Elevated +++++
Elevated +
Níveis de cistina lasma após um teste de carga oral
Same
Same ou ligeiro aumento
Increased
Transporte intestinal
Absent (sem transporte de cistina, Lisina, ou arginina)
Absent
Reduzido
Provas recentes sugerem que as contas complexas 4F2HC/4F2LC para o sistema de transporte de aminoácidos Y+L na superfície basolateral das células intestinais e proximais renais tubulares e que as mutações do gene 4F2LC (SLC7A7) na banda 14q11-13 causam a rara doença recessiva chamada intolerância à lisina-proteína.
Sumário
rBAT, uma glicoproteína 90-kd tipo II, é um transportador de alta afinidade, independente do sódio, para os aminoácidos dibásicos nos túbulos renais convoluídos proximais em roedores. Curiosamente, a análise de ligação sugere que esta é a mesma região para a qual um locus cistinúrico, SLC3A1, foi identificado.160 mutações diferentes no gene SLC3A1 e 116 no gene SLC7A9 foram identificadas em pacientes com cistinúria em todo o mundo.
Cistinúria dos tipos III e II (não tipo I) foram ligadas à banda 19p13.1 (SLC7A9); entretanto, estudos adicionais são necessários para determinar o papel exato do gene SLC7A9. Aproximadamente 50% das crianças com 2 mutações SLC3A1 (cistinúria homozigotos clássica tipo I) desenvolvem pelo menos uma pedra dentro da primeira década de vida.