Transportul renal al cistinei
Aminoacizii sunt ușor filtrați de glomerul și sunt supuși unei reabsorbții aproape complete de către celulele tubulare proximale. Doar 0,4% din cistina filtrată apare în urină. Diverși autori au studiat transportul aminoacizilor în membranele celulare obținute din tubulii renali proximali ai oamenilor, șobolanilor și iepurilor. Cel puțin 2 sisteme de transport sunt responsabile de reabsorbția cistinei, după cum urmează:
-
Sistem de înaltă afinitate: Acest sistem este afectat la persoanele cu cistinurie. Sistemul de înaltă afinitate mediază absorbția a 10% din L-cistină și a aminoacizilor dibazici la nivelul membranei apicale a celui de-al treilea segment drept (S3) al tubului proximal.
-
Sistemul de joasă afinitate: Acest sistem este prezent în partea S1-S2 a tubului proximal și este responsabil pentru 90% din reabsorbția L-cistinei. Procesul de joasă afinitate sporește procesul de înaltă afinitate. După absorbție, fiecare moleculă de cistină este transformată intracelular în 2 molecule de cisteină. Cisteina iese prin membrana bazolaterală.
Studiile genetice ale ADN-ului din familiile cu cistinurie relevă o genă defectuoasă localizată pe cromozomul 2. Gena care codifică transportorul de cistină, denumită inițial rBAT, este cunoscută acum ca SLC3A1 (SLC pentru transportor de solut) în baza de date internațională a genomului. O a doua genă a cistinuriei, situată pe cromozomul 19, se numește SLC7A9. Gena SLC7A9 normală codifică o subunitate a transportatorului de cistină numită b 0,+ AT (transportator de aminoacizi). Procesul de absorbție a cistinei este activat de produsele genelor SLC3A1 și SLC7A9. Transportul de L-cistină în vezicula membranară de la marginea perilor este independent de sodiu și electrogen. La persoanele cu cistinurie, mișcarea cistinei sau a cisteinei din celulele tubulare în sânge nu este afectată.
Transportul intestinal al cistinei
Transportorul de înaltă afinitate este prezent în membrana apicală de tip brush-border a jejunului și este responsabil pentru absorbția cistinei și a aminoacizilor dibazici. Majoritatea pacienților cu cistinurie au o absorbție deficitară a cistinei; cu toate acestea, deficitul de cistină nu este semnificativ din punct de vedere clinic deoarece absorbția aminoacizilor cu lanț scurt nu este afectată.
În mod normal, cistina și ceilalți aminoacizi dibazici (adică ornitina, lizina, arginina) sunt filtrați la nivelul glomerulului și reabsorbiți în tubul convolutiv proximal printr-un canal transmembranar luminal de înaltă afinitate. Defectele acestui canal determină niveluri ridicate de secreție de aminoacizi dibazici în urină. În timp ce ornitina, lizina și arginina sunt complet solubile, cistina este relativ insolubilă la niveluri fiziologice ale pH-ului urinar de 5-7, cu un nivel pKa de 8,3. La un nivel al pH-ului urinar de 7,8 și 8, solubilitatea respectivă a cistinei este aproape dublată și triplată.
Dent și Senior au demonstrat că solubilitatea cistinei este dependentă de pH. Solubilitatea cistinei în urină este de aproximativ 250 mg/L (1 mmol/L) până la un nivel al pH-ului de 7, dar solubilitatea crește cu un nivel mai ridicat al pH-ului cu până la 500 mg/L (2 mmol/L) sau mai mult peste un nivel al pH-ului de 7,5, așa cum este descris în imaginea de mai jos. Măsurătorile în urină au arătat în mod clar că solubilitatea cistinei crește liniar odată cu creșterea puterii ionice. Pak și colegii săi au arătat că aproximativ 70 mg de cistină suplimentară pot fi dizolvate în fiecare litru de soluție, cu o creștere a tăriei ionice de la 0,005-0,3. În plus, la aceeași tărie ionică și același pH, solubilitatea cistinei variază în funcție de tipul special de electrolit prezent. A se vedea imaginea de mai jos.
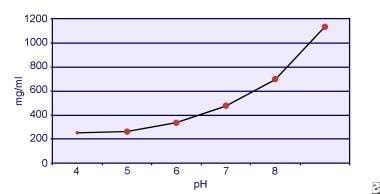 Solubilitatea cistinei în urină.
Solubilitatea cistinei în urină. Experimentele in vitro efectuate de Pak și Fuller în 1983 au arătat că cea mai mare solubilitate se realizează în prezența clorurii de calciu, urmată de clorura de magneziu și clorura de sodiu. Mai mult, solubilitatea cistinei este, de asemenea, afectată de macromoleculele urinare. S-a demonstrat că prezența coloidului în urina normală crește solubilitatea cistinei; cu toate acestea, mecanismul acestei acțiuni nu este clar. Deoarece nimic nu inhibă cristalizarea cistinei, principalul factor determinant este suprasaturația urinară. Nuclearea eterogenă a oxalatului de calciu, a brushitului sau a hidroxiapatitei nu apare la indivizii cu cistinurie.
Factorii de risc pentru cristalizarea cistinei includ (1) un nivel scăzut al pH-ului, (2) o rezistență ionică redusă, (3) prezența cristalelor de cistină și (4) niveluri scăzute de macromolecule urinare.
Cistina este un homodimer al cisteinei legat prin disulfură și are următoarea structură:
COOH-CHNH2-CH2 -S-S-CH2-CHNH2- COOH Cistină
COOH-CHNH2-CH2 -SH Cisteină
Cistina este absorbită în intestinul subțire într-un mod similar cu cel al rinichilor. La persoanele cu cistinurie, absorbția intestinală a cistinei este, de asemenea, afectată în grade diferite. Metabolismul metioninei este o altă sursă de cistină serică. Două domenii de glicoproteine membranare de tip II au fost implicate în transportul aminoacizilor prin membrana plasmatică. Primul este rBAT, iar al doilea este 4F2HC (lanțul greu al antigenului 4F2)
Două treimi din persoanele cu cistinurie care formează calculi fac calculi de cistină pură, iar o treime au un amestec de calculi de cistină și oxalat de calciu. În 2002, Martins et al. au raportat că precipitarea oxalatului de calciu are loc printr-un proces de sărare, adică reducerea solubilității unei substanțe datorită adaosului unei alte substanțe în sistem, mai degrabă decât prin procesul de nucleare eterogenă. Hipocitraturia, hipercalciuria și hiperuricosuria sunt, de asemenea, frecvent asociate cu cistinuria. Având în vedere structura lor cristalină relativ uniformă, fără planuri de clivaj lamelare, calculii de cistină pură sunt printre cei mai duri pe indicele de fragilitate a calculilor lui Dretler.
Cistinuria este o boală autosomal-recesivă împărțită în 3 subtipuri: Rosenberg I, II și III. Cistinuria de tip I este cea mai frecventă variantă și a fost cartografiată la banda 2p16.3. Heterozigoții de tip I prezintă o aminoacidurie normală. S-a crezut că cistinuria clasică, tipurile II și III, sunt variante alelice, dar analizele de legătură au arătat că tipul III este un defect al unei gene necaracterizate (SLC7A9) pe banda 19q13.1. Heterozigoții tipurilor II și III manifestă adesea cistinurie fără calculi de cistină și pot avea un risc crescut pentru alte tipuri de urolitiază. Heterozigoții de tip I se disting prin niveluri normale de cistină urinară.
În comparație cu homozigoții de tip I și II, homozigoții de tip III prezintă o creștere a concentrației plasmatice de cistină după administrarea orală de cistină. Harris și colab. au raportat natura complexă a geneticii cistinuriei prin măsurarea nivelului de excreție a cistinei urinare la părinții (heterozigoți obligatorii) ai probandelor de cistinurie și au descoperit alele complet recesive (ambii părinți excretau cistină în intervalul de referință) și alele dominante (ambii părinți excretau cistină la niveluri ridicate).
Pentru clasificarea clinică a cistinuriei, cistina urinară poate fi măsurată la fiecare părinte al unui proband ca fenotip I (recesiv, nivel de cistină urinară < 100 µmol/g de creatinină), fenotip II (dominant, nivel de cistină urinară >1000 µmol/g de creatinină) și fenotip III (parțial dominant, nivel de cistină urinară 100-1000 µmol/g de creatinină). Cistinuria poate fi, de asemenea, clasificată pe baza vârstei la care apar primele simptome (adică, infantilă, juvenilă, adolescentină).
La indivizii sănătoși, limita superioară de excreție a cistinei este de 20 mg/g de creatinină (< 10 µmol/mmol de creatinină). Homozigoții excretă mai mult de 400 mg/zi (1,7 mmol/zi), iar excreția de cistină la pacienții homozigoți este de obicei de 600-1400 mg/zi (2,5-5,8 mmol/zi). Heterozigoții cu cistinurie de tip I și III excretă mai puțin de 200 mg/zi (0,8 mmol/d) și nu formează calculi. Heterozigoții de tip II excretă până la 200-400 mg/zi, dar acești pacienți pot forma calculi. Incidența formării de calculi crește atunci când concentrația de cistină urinară depășește 700 µmol/L (170 mg/L).
Genetică
În ultimii ani, odată cu progresele în biologia moleculară, s-au acumulat noi informații privind fiziopatologia cistinuriei. În 1992, mai mulți cercetători au raportat clonarea expresiei unui ADNc renal de 2,3 kilobaze (D2H sau rBAT) care a indus captarea independentă de sodiu a cistinei și a aminoacizilor dibazici în ovocitele Xenopus laevis injectate cu ARNc. Gena rBAT a fost cartografiată pe cromozomul 2 (banda 2p21) între D2S119 și D2S288. Această genă se numește acum SLC3A1 în baza de date a genomului.
Studiile imunohistochimice și de hibridizare in situ au arătat că rBAT este exprimată în celulele segmentului S3 (pars recta) al tubului proximal și al intestinului subțire, la nivelul membranei luminale a marginii în perie. În 1995, Gasparini et al. au raportat că mutațiile în SLC3A1 au apărut la pacienții cu cistinurie de tip I și nu la pacienții cu cistinurie de tip II sau III. Până în prezent, au fost descrise mai mult de 160 de mutații diferite, incluzând atât deleții mici, cât și mari de perechi de baze ADN din genă. Una dintre cele mai frecvente modificări genetice în SLC3A1 se numește M467T, iar majoritatea mutațiilor tind să fie specifice unei populații. Mutația M467T este destul de frecventă în populațiile mediteraneene. Interesant este faptul că a reprezentat 40% din mutații într-o cohortă spaniolă de familii și a fost rară la pacienții studiați în Quebec, Canada.
În 1999, a fost izolată gena SLC7A9 (BAT1). Gena codifică o proteină de 487 aminoacizi și a fost cartografiată pe cromozomul 19 (banda 19q13) între D19S414 și D19S220. Produsul BAT1 pare a fi o proteină de membrană cu 12 regiuni care se întind pe membrană. Mutațiile în gena BAT1 cauzează probabil cistinuria non-tip I (tipul II și III Rosenberg). Mutațiile la nivelul locusului 19q sunt deosebit de frecvente în rândul evreilor libieni, iar riscul de urolitiază la pacienții care moștenesc 2 astfel de mutații la nivelul locusului 19q este aproximativ comparabil cu cel al pacienților care moștenesc 2 mutații rBAT.
116 mutații în această genă au fost raportate. Cea mai frecventă mutație la evreii libieni a avut ca rezultat o metionină care înlocuiește reziduul de aminoacid valină (V170M) în proteină. La heterozigoții cu mutația V170M, concentrațiile urinare de cistină variază între 86-1238 µmol/g de creatinină. Astfel, unele dintre valorile la heterozigoții V170M sunt în concordanță cu cistinuria de tip III, iar altele cu cistinuria de tip II.
O caracteristică distinctivă aparentă între cistinuria de tip II și cistinuria de tip III este lipsa absorbției intestinale a cistinei la homozigoții de tip II. În 2000, Pras a sugerat o nouă clasificare bazată pe analiza moleculară. Recent, Dello Strologo et al. au propus o nouă clasificare genetică, după cum urmează:
-
Tip A, mutație a ambelor alele ale SLC3A1: Heterozigoții prezintă un model urinar normal de aminoacizi.
-
Tip B, mutație a ambelor alele ale SLC7A9: Heterozigoții prezintă de obicei o creștere a excreției urinare de cistină și aminoacizi dibazici.
-
Tip AB, cistinurie cauzată de 1 mutație în SLC3A1 și 1 mutație în SLC7A9: Cistinuria de tip mixt poate fi cauzată de interacțiunea a 2 gene mutante distincte, iar proteina codificată de gena 19q interacționează direct cu rBAT în segmentul S3 al tubului proximal (a se vedea tabelul).
Martens et al. (2008) au raportat recent 3 sindroame de deleție genetică asociate cu cistinuria de tip A: sindromul de deleție 2p21, sindromul de hipotonie-cistinurie (HCS) și o formă atipică a sindromului de hipotonie-cistinurie. Ambele alele SLC3A1 și PREPL lipsesc la pacienții cu HCS. O genă suplimentară (C2orf34) este ștearsă în cazul HCS atipic.
Tabel. Clasificarea cistinuriei (Open Table in a new window)
|
Rosenberg et al |
Tipul I |
Tipul II |
Tipul III |
|
Tipul I |
Tip I |
Nu de tip I |
|
|
Gena responsabilă |
SLC3A1 |
SLC7A9 |
|
|
Bandă |
2p21 |
19q13.1 |
|
|
Nr. de mutații |
>60 |
||
|
Mutația cea mai frecventă |
M467 |
V170M |
|
|
Populația afectată |
Mediteraneeni spanioli, 40% |
Evrei libieni |
|
|
Rata de ștergere |
54% |
25% |
|
|
Proteine |
rBAT |
BAT1 |
|
|
Sistem de transport al aminoacizilor |
|||
|
Localizare în tubulul convertit proximal |
S3 |
S1, S2 |
|
|
Caracteristică de transport |
Afinitate ridicată, capacitate scăzută |
Afinitate scăzută, capacitate mare |
|
|
Caracteristică clinică |
|||
|
Homozigoți |
Simptomatici |
aproximativ 90% simptomatici |
|
|
Heterozigoți |
Asimptomatici |
aproximativ 10%-13% simptomatici |
|
|
Nivelul de cistină urinară |
Normal |
Elevat +++++ |
Elevat + |
|
Nivelul de cistină în plasmă după un test de încărcare pe cale orală |
Identic |
Identic sau creștere ușoară |
Creștere |
|
Transport intestinal |
Absent (nu există transport de cistină, lizină, sau arginină) |
Absent |
Redus |
Potriviri recente sugerează că complexul 4F2HC/4F2LC reprezintă pentru sistemul de transport al aminoacizilor Y+L la suprafața bazolaterală a celulelor tubulare proximale intestinale și renale și că mutațiile genei 4F2LC (SLC7A7) de pe banda 14q11-13 determină boala recesivă rară numită intoleranță la proteinele de lizină.
Rezumat
rBAT, o glicoproteină de tip II de 90 kd, este un transportor de mare afinitate, independent de sodiu pentru aminoacizii dibazici în tubulii renali convoluți proximali la rozătoare.Gena rBAT umană a fost localizată pe banda 2p21. În mod interesant, analiza de legătură sugerează că aceasta este aceeași regiune în care a fost identificat un locus cistinuric, SLC3A1.160 de mutații diferite în gena SLC3A1 și 116 în gena SLC7A9 au fost identificate la pacienții cu cistinurie din întreaga lume.
Cistinuria de tip III și II (non-tip I) au fost legate de banda 19p13.1 (SLC7A9); cu toate acestea, sunt necesare studii suplimentare pentru a determina rolul exact al genei SLC7A9. Aproximativ 50% dintre copiii cu 2 mutații SLC3A1 (cistinuria clasică homozigotă de tip I) dezvoltă cel puțin un calcul în primul deceniu de viață.
.