Renal transport av cystin
Aminosyror filtreras lätt av glomerulus och genomgår nästan fullständig reabsorption av proximala tubulära celler. Endast 0,4 % av det filtrerade cystinet förekommer i urinen. Olika författare har studerat aminosyratransporten i cellmembran som erhållits från den proximala njurtubuli från människor, råttor och kaniner. Minst 2 transportsystem är ansvariga för cystinreabsorptionen enligt följande:
-
Högaffinitetssystem: Detta system är påverkat hos personer med cystinuri. Systemet med hög affinitet medierar upptag av 10 % L-cystin och de dibasiska aminosyrorna vid det apikala membranet i det raka tredje segmentet (S3) i den proximala tubuli.
Låg affinitetssystem: Detta system finns i S1-S2-delen av proximala tubuli och är ansvarigt för 90 % av L-cystinreabsorptionen. Processen med låg affinitet förstärker processen med hög affinitet. Efter absorptionen omvandlas varje cystinmolekyl intracellulärt till två cysteinmolekyler. Cystein lämnar ut vid det basolaterala membranet.
Genetiska studier av DNA från familjer med cystinuri avslöjar en defekt gen som är belägen på kromosom 2. Den gen som kodar för cystintransportören, som ursprungligen kallades rBAT, är nu känd som SLC3A1 (SLC för solute carrier) i den internationella Genome Database. En andra cystinuriagene på kromosom 19 kallas SLC7A9. Den normala SLC7A9-genen kodar för en underenhet av cystintransportören som kallas b 0,+ AT (aminosyratransportör). Processen för cystinupptag aktiveras av genprodukterna SLC3A1 och SLC7A9. Transporten av L-cystin i borstgränsmembranvesikeln är natriumoberoende och elektrogen. Hos personer med cystinuri är förflyttningen av cystin eller cystein från de tubulära cellerna till blodet inte påverkad.
Intestinal transport av cystin
Transportören med hög affinitet finns i det apikala borstgränsmembranet i jejunum och är ansvarig för absorptionen av cystin och dibasiska aminosyror. De flesta patienter med cystinuri har nedsatt absorption av cystin; cystinbrist är dock inte kliniskt signifikant eftersom absorptionen av kortkedjiga aminosyror inte påverkas.
Normalt filtreras cystin och de andra dibasiska aminosyrorna (dvs. ornitin, lysin, argininin) vid glomerulus och återabsorberas i den proximala konvoluterade tubulären genom en luminal transmembrankanal med hög affinitet. Defekter i denna kanal orsakar förhöjda nivåer av dibasiska aminosyror i urinen. Ornitin, lysin och arginin är fullständigt lösliga, medan cystin är relativt olösligt vid fysiologiska pH-nivåer i urinen på 5-7, med en pKa-nivå på 8,3. Vid ett pH-värde i urinen på 7,8 och 8 fördubblas respektive tredubblas cystins löslighet nästan.
Dent och Senior visade att cystins löslighet är pH-beroende. Lösligheten av cystin i urin är cirka 250 mg/L (1 mmol/L) upp till en pH-nivå på 7, men lösligheten ökar med en högre pH-nivå med upp till 500 mg/L (2 mmol/L) eller mer över en pH-nivå på 7,5, vilket framgår av bilden nedan. Mätningar i urin har tydligt visat att cystinlösligheten ökar linjärt med ökad jonstyrka. Pak och medarbetare visade att cirka 70 mg ytterligare cystin kan lösas upp i varje liter lösning, med en ökning av jonstyrkan från 0,005-0,3. Dessutom varierar cystinlösligheten vid samma jonstyrka och pH-värde beroende på vilken typ av elektrolyt som används. Se bilden nedan.
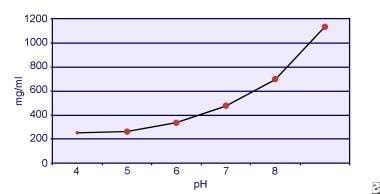 Cystinlöslighet i urin.
Cystinlöslighet i urin. In vitro-experiment av Pak och Fuller 1983 visade att den högsta lösligheten uppnås i närvaro av kalciumklorid, följt av magnesium- och natriumklorid. Vidare påverkas cystinlösligheten även av makromolekyler i urinen. Närvaron av kolloid i normal urin har visat sig öka cystinlösligheten, men mekanismen för denna verkan är inte klarlagd. Eftersom ingenting hämmar cystinkristalliseringen är den viktigaste faktorn övermättnad i urinen. Heterogen kärnbildning av kalciumoxalat, brushit eller hydroxyapatit förekommer inte hos personer med cystinuri.
Riskfaktorer för cystinkristallisation är (1) låg pH-nivå, (2) minskad jonstyrka, (3) förekomst av cystinkristaller och (4) låga halter av makromolekyler i urinen.
Cystin är en disulfidbunden homodimer av cystein och har följande struktur:
COOH-CHNH2-CH2 -S-S-CH2-CHNH2- COOH Cystin
COOH-CHNH2-CH2-CH2 -SH Cystein
Cystin absorberas i tunntarmen på ett sätt som liknar njurarnas. Hos personer med cystinuri är tarmabsorptionen av cystin också nedsatt i varierande grad. Metabolism av metionin är en annan källa till serumcystin. Två membranglykoproteindomäner av typ II har involverats i aminosyratransporten via plasmamembranet. Den första är rBAT och den andra är 4F2HC (den tunga kedjan av 4F2-antigenet)
Två tredjedelar av personer med cystinuri som bildar stenar gör rena cystinstenar och en tredjedel har en blandning av cystin- och kalciumoxalatstenar. År 2002 rapporterade Martins et al att utfällning av kalciumoxalat sker genom en saltningsprocess, dvs. minskad löslighet hos en substans på grund av tillsats av en annan substans i systemet, snarare än genom processen med heterogen kärnbildning. Hypokitraturi, hyperkalciuri och hyperurikosuri är också ofta förknippade med cystinuri. Med tanke på deras relativt enhetliga kristallina struktur utan lamellformade klyvningsytor är rena cystinstenar bland de hårdaste på Dretlers stenfragilitetsindex.
Cystinuri är en autosomalt recessiv sjukdom som är uppdelad i tre subtyper: Rosenberg I, II och III. Cystinuri typ I är den vanligaste varianten och har kartlagts till band 2p16.3. Heterozygoter av typ I uppvisar normal aminoaciduri. Klassisk cystinuri, typ II och III, ansågs vara alleliska varianter, men kopplingsanalyser har visat att typ III är en defekt i en okaraktäriserad gen (SLC7A9) på band 19q13.1. Heterozygoter av typ II och III uppvisar ofta cystinuri utan cystinstenar och kan ha en ökad risk för andra typer av urolithiasis. Heterozygoter av typ I kännetecknas av normala nivåer av cystin i urinen.
I motsats till homozygoter av typ I och typ II uppvisar homozygoter av typ III en ökning av plasmakoncentrationen av cystin efter oral administrering av cystin. Harris et al rapporterade den komplexa karaktären hos genetiken för cystinuri genom att mäta nivån av cystinutsöndring i urinen hos föräldrarna (obligatoriska heterozygoter) till cystinuri-proband och fann helt recessiva alleler (båda föräldrarna utsöndrade cystin inom referensområdet) och dominanta alleler (båda föräldrarna utsöndrade cystin på höga nivåer).
För att klassificera cystinuri kliniskt kan urincystin mätas hos varje förälder till en proband som fenotyp I (recessiv, urincystinnivå < 100 µmol/g kreatinin), fenotyp II (dominant, urincystinnivå >1000 µmol/g kreatinin) och fenotyp III (delvis dominant, urincystinnivå 100-1000 µmol/g kreatinin). Cystinuri kan också klassificeras utifrån den ålder då symtomen först uppträder (dvs. infantil, juvenil, tonåring).
I friska individer är den övre gränsen för cystinutsöndring 20 mg/g kreatinin (< 10 µmol/mmol kreatinin). Homozygoter utsöndrar mer än 400 mg/d (1,7 mmol/d), och cystinutsöndringen hos homozygota patienter är vanligtvis 600-1400 mg/d (2,5-5,8 mmol/d). Heterozygoter med cystinuri av typ I och III utsöndrar mindre än 200 mg/d (0,8 mmol/d) och bildar inte stenar. Heterozygoter av typ II utsöndrar upp till 200-400 mg/d, men dessa patienter kan bilda stenar. Förekomsten av stenbildning ökar när cystinkoncentrationen i urinen överstiger 700 µmol/L (170 mg/L).
Genetik
Under de senaste åren, med framstegen inom molekylärbiologin, har nya insikter ackumulerats när det gäller cystinuriens patofysiologi. År 1992 rapporterade flera forskare om uttryckskloning av ett 2,3 kilobas renalt cDNA (D2H eller rBAT) som inducerade ett natriumoberoende upptag av cystin och dibasiska aminosyror i cRNA-injicerade Xenopus laevis oocyter. rBAT-genen kartlades på kromosom 2 (band 2p21) mellan D2S119 och D2S288. Denna gen heter nu SLC3A1 i Genome Database.
Immunohistokemiska och in situ-hybridiseringsstudier visade att rBAT uttrycks i celler i S3-segmentet (pars recta) av proximala tubuli och tunntarmen vid det luminala borstgränsmembranet. År 1995 rapporterade Gasparini et al att mutationer i SLC3A1 förekom hos patienter med cystinuri av typ I och inte hos patienter med cystinuri av typ II eller III. Hittills har mer än 160 olika mutationer beskrivits, inklusive både små och stora deletioner av DNA-baspar från genen. En av de vanligaste genetiska förändringarna i SLC3A1 kallas M467T, och de flesta mutationer tenderar att vara populationsspecifika. M467T-mutationen är ganska vanlig i Medelhavspopulationer. Intressant nog stod den för 40 % av mutationerna i en spansk kohort av familjer och var sällsynt hos patienter som studerades i Quebec, Kanada.
År 1999 isolerades genen SLC7A9 (BAT1). Genen kodar för ett protein med 487 aminosyror och kartlades till kromosom 19 (band 19q13) mellan D19S414 och D19S220. BAT1-produkten verkar vara ett membranprotein med 12 membranöverskridande regioner. Mutationer i BAT1-genen orsakar troligen cystinuri som inte är av typ I (Rosenberg typ II och III). Mutationer på lokus 19q är särskilt vanliga bland libyska judar, och risken för urolithiasis hos patienter som ärver två sådana mutationer på lokus 19q är ungefär jämförbar med risken hos patienter som ärver två rBAT-mutationer.
116 mutationer i denna gen har rapporterats. Den vanligaste mutationen hos libyska judar resulterade i att ett metionin ersatte aminosyraresten valin (V170M) i proteinet. Hos heterozygoter med V170M-mutationen varierar cystinkoncentrationerna i urinen från 86-1238 µmol/g kreatinin. Vissa av värdena hos V170M-heterozygoter överensstämmer således med cystinuri av typ III och andra med cystinuri av typ II.
Ett uppenbart särskiljande kännetecken mellan cystinuri av typ II och typ III är avsaknaden av intestinal cystinabsorption hos homozygoter av typ II. År 2000 föreslog Pras en ny klassificering baserad på molekylär analys. Nyligen har Dello Strologo et al. föreslagit en ny genetisk klassificering enligt följande:
-
Typ A, mutation av båda allelerna i SLC3A1: Heterozygoter uppvisar ett normalt aminosyraurinmönster.
-
Typ B, mutation av båda allelerna i SLC7A9: Heterozygoter uppvisar vanligen en ökning av cystin- och dibasisk aminosyraurinutsöndringen.
-
Typ AB, cystinuri orsakad av 1 mutation i SLC3A1 och 1 mutation i SLC7A9: Cystinuri av blandad typ kan orsakas av interaktionen mellan 2 olika muterade gener, och proteinet som kodas av 19q-genen interagerar direkt med rBAT i S3-segmentet i den proximala tubulären (se tabell).
Martens et al (2008) rapporterade nyligen 3 gen-deletionssyndrom som är associerade med cystinuri av typ A: 2p21-deletionssyndromet, hypotoni-cystinuriasyndromet (HCS) och en atypisk form av hypotoni-cystinuriasyndromet. Båda allelerna av SLC3A1 och PREPL saknas hos patienter med HCS. Ytterligare en gen (C2orf34) är borttagen vid atypisk HCS.
Tabell. Klassificering av cystinuri (Öppna tabellen i ett nytt fönster)
Rosenberg et al
Typ I
Typ II
Typ III
Molekylär
Typ I
Inte typ I
Respektive gen
SLC3A1
SLC7A9
Band
2p21
19q13.1
Nr. mutationer
>60
Första förekommande mutation
M467
V170M
Population som berörs
Medelhavssvenskar, 40%
Libyska judar
Deletionsfrekvens
54%
25%
Protein
rBAT
BAT1
Aminosyretransportsystem
Lokalisering i proximala konverterade tubuli
S3
S1, S2
Transportörens egenskaper
Hög affinitet, låg kapacitet
Låg affinitet, hög kapacitet
Kliniska kännetecken
Homozygoter
Symtomatiska
Omkring 90 % symtomatiska
Heterozygoter
Asymptomatiska
Omkring 10 %-13% symtomatiska
Urinära cystinnivåer
Normala
Normala +++++
Normala +
Plasma cystinnivåer efter ett oralt belastningstest
Samma
Samma eller liten ökning
Ökat
Intestinal transport
Absent (ingen transport av cystin, lysin, eller arginin)
Absent
Reducerad
Nyligen framkomna bevis tyder på att 4F2HC/4F2LC-komplexet står för för transportsystemet för aminosyror av typen Y+L på den basolaterala ytan av proximala tubulära celler i tarmen och njurarna och att mutationer i 4F2LC-genen (SLC7A7) på band 14q11-13 orsakar den sällsynta recessiva sjukdomen lysinproteinintolerans.
Sammanfattning
rBAT, ett 90 kd typ II-glykoprotein, är en natriumoberoende transportör med hög affinitet för dibasiska aminosyror i de proximala konvoluterade njurtubuli hos gnagare. rBAT-genen från människa har lokaliserats på band 2p21. Intressant nog tyder kopplingsanalysen på att detta är samma region till vilken ett cystinuriskt locus, SLC3A1, har identifierats.160 olika mutationer i SLC3A1-genen och 116 i SLC7A9-genen har identifierats hos patienter med cystinuri i hela världen.
Typ III och II cystinuri (ej typ I) har kopplats till band 19p13.1 (SLC7A9), men ytterligare studier behövs för att fastställa SLC7A9-genens exakta roll. Ungefär 50 % av barnen med 2 SLC3A1-mutationer (klassisk homozygot typ I-cystinuri) utvecklar minst en sten inom det första decenniet av livet.
 Cystinlöslighet i urin.
Cystinlöslighet i urin.