Kystiinin renaalinen kuljetus
Aminohapot suodattuvat helposti glomeruluksessa, ja ne imeytyvät lähes täydellisesti proksimaalisten tubulussolujen kautta. Vain 0,4 % suodatetusta kystiinistä päätyy virtsaan. Useat kirjoittajat ovat tutkineet aminohappojen kulkeutumista solukalvoissa, jotka on saatu ihmisten, rottien ja kanien proksimaalisesta munuaistubuluksesta. Kystiinin takaisinimeytymisestä vastaa ainakin 2 kuljetusjärjestelmää, jotka ovat seuraavat:
-
High-affinity system: Tämä järjestelmä vaikuttaa henkilöillä, joilla on kystinuria. Korkea-affiniteettijärjestelmä välittää 10 %:n L-kystiinin ja dibasisten aminohappojen imeytymistä proksimaalisen tubuluksen suoran kolmannen segmentin (S3) apikaalisella kalvolla.
Matala-affiniteettijärjestelmä: Tämä järjestelmä on läsnä proksimaalisen tubuluksen S1-S2-osassa ja vastaa 90 %:sta L-kystiinin reabsorptiosta. Matalan affiniteetin prosessi lisää korkean affiniteetin prosessia. Imeytymisen jälkeen jokainen kystiinimolekyyli muunnetaan solunsisäisesti kahdeksi kysteiinimolekyyliksi. Kysteiini poistuu basolateraalikalvolla.
Kystinuriaa sairastavien perheiden DNA:n geneettiset tutkimukset paljastavat viallisen geenin, joka sijaitsee kromosomissa 2. Kystiinikuljettajaa koodaava geeni, jota alun perin kutsuttiin nimellä rBAT, tunnetaan nykyään kansainvälisessä genomitietokannassa nimellä SLC3A1 (SLC for solute carrier). Toinen kromosomissa 19 oleva kystinurian geeni on nimeltään SLC7A9. Normaali SLC7A9-geeni koodaa kystiinikuljettajan alayksikköä nimeltä b 0,+ AT (aminohappokuljettaja). SLC3A1- ja SLC7A9-geenien tuotteet aktivoivat kystiinin ottoprosessin. L-kystiinin kuljetus harjan reunakalvon vesikkelissä on natriumista riippumatonta ja elektrogeenistä. Henkilöillä, joilla on kystinuria, kystiinin tai kysteiinin siirtyminen tubulussoluista vereen ei vaikuta.
Kystiinin suolistokuljetus
Korkea-affiniteettikuljettaja on jejunumin apikaalisessa harjasreunakalvossa ja vastaa kystiinin ja dibasisten aminohappojen imeytymisestä. Useimmilla kystinuriapotilailla kystiinin imeytyminen on heikentynyt; kystiinin puutos ei kuitenkaan ole kliinisesti merkittävä, koska lyhytketjuisten aminohappojen imeytyminen ei vaikuta.
Normaalisti kystiini ja muut dibasiset aminohapot (ts. ornitiini, lysiini, arginiini) suodattuvat glomeruluksessa ja imeytyvät takaisin proksimaaliseen konvoluutiomaiseen tubulukseen korkea-affiniteettisen lumaalisen transmembraanisen kanavan kautta. Tämän kanavan viat aiheuttavat kohonneita dibaasisten aminohappojen eritystä virtsaan. Ornitiini, lysiini ja arginiini ovat täysin liukoisia, mutta kystiini on suhteellisen liukenematon virtsan fysiologisessa pH-tasossa 5-7, ja sen pKa-taso on 8,3. Virtsan pH-tasoilla 7,8 ja 8 kystiinin vastaava liukoisuus lähes kaksinkertaistuu ja kolminkertaistuu.
Dent ja Senior osoittivat, että kystiinin liukoisuus on pH-riippuvainen. Kystiinin liukoisuus virtsaan on noin 250 mg/l (1 mmol/l) pH-tasoon 7 asti, mutta liukoisuus kasvaa pH-tason noustessa jopa 500 mg/l (2 mmol/l) tai enemmän pH-tason 7,5 yläpuolella, kuten alla olevassa kuvassa on esitetty. Virtsasta tehdyt mittaukset ovat selvästi osoittaneet, että kystiinin liukoisuus kasvaa lineaarisesti ionivahvuuden kasvaessa. Pak ja kollegat osoittivat, että jokaiseen litraan liuosta voidaan liuottaa noin 70 mg lisää kystiiniä, kun ionivahvuus kasvaa 0,005-0,3:sta. Lisäksi kystiinin liukoisuus vaihtelee samalla ionivahvuudella ja pH:lla sen mukaan, minkä tyyppisestä elektrolyytistä on kyse. Katso alla oleva kuva.
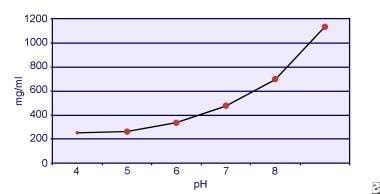 Kystiinin liukoisuus virtsaan.
Kystiinin liukoisuus virtsaan. Pakin ja Fullerin vuonna 1983 tekemät in vitro -kokeet osoittivat, että suurin liukoisuus saavutetaan kalsiumkloridin läsnäollessa, jonka jälkeen tulevat magnesium- ja natriumkloridi. Lisäksi kystiinin liukoisuuteen vaikuttavat myös virtsan makromolekyylit. Kolloidin läsnäolon normaalissa virtsassa on osoitettu lisäävän kystiinin liukoisuutta; tämän vaikutuksen mekanismi ei kuitenkaan ole selvä. Koska mikään ei estä kystiinin kiteytymistä, tärkein tekijä on virtsan ylikylläisyys. Kalsiumoksalaatin, brushiitin tai hydroksiapatiitin heterogeenista ydintymistä ei tapahdu henkilöillä, joilla on kystinuria.
Riskitekijöitä kystiinin kiteytymiselle ovat (1) matala pH-taso, (2) alentunut ionivoimakkuus, (3) kystiinikiteiden esiintyminen ja (4) virtsan matalat makromolekyylitasot.
Kystiini on kysteiinin disulfidisidoksinen homodimeeri ja sillä on seuraava rakenne:
COOH-CHNH2-CH2 -S-S-S-CH2-CHNH2- COOH Kystiini
COOH-CHNH2-CH2 -SH Kysteiini
Kystiini imeytyy ohutsuolesta munuaisten tapaan. Henkilöillä, joilla on kystinuria, myös kystiinin imeytyminen suolistosta on vaihtelevassa määrin heikentynyt. Metioniinin aineenvaihdunta on toinen seerumin kystiinin lähde. Kahden tyypin II kalvoglykoproteiinidomeenin on todettu osallistuvan aminohappojen kuljetukseen plasmakalvon kautta. Ensimmäinen on rBAT ja toinen on 4F2HC (4F2-antigeenin raskas ketju)
Kahdella kolmasosalla henkilöistä, joilla on kystinuria ja jotka muodostavat kiviä, on puhtaita kystiinikiviä, ja kolmasosalla on kystiini- ja kalsiumoksalaattikivien sekoitus. Vuonna 2002 Martins ym. raportoivat, että kalsiumoksalaatin saostuminen tapahtuu pikemminkin salting-out-prosessin eli aineen liukoisuuden vähenemisen kautta, joka johtuu toisen aineen lisäämisestä systeemiin, kuin heterogeenisen ydintymisprosessin kautta. Hypokitraturia, hyperkalsiuria ja hyperurikosuria liittyvät usein myös kystinuriaan. Ottaen huomioon suhteellisen yhtenäisen kiderakenteensa, jossa ei ole lamellimaisia pilkkoutumistasoja, puhtaat kystiinikivet kuuluvat Dretlerin kiven haurausindeksin kovimpiin.
Kystinuria on autosomaalisesti resessiivinen sairaus, joka jakautuu kolmeen alatyyppiin: Rosenberg I, II ja III. Kystinuria tyyppi I on yleisin variantti, ja se on kartoitettu kaistalle 2p16.3. Tyypin I heterotsygooteilla on normaali aminohappoasiduria. Klassista kystinuriaa, tyyppejä II ja III, pidettiin allelivariantteina, mutta kytkentäanalyysit ovat paljastaneet, että tyyppi III on luonnehtimattoman geenin (SLC7A9) vika kaistalla 19q13.1. Tyypin II ja III heterotsygooteilla esiintyy usein kystinuriaa ilman kystiinikiviä, ja heillä saattaa olla suurentunut riski sairastua muihin virtsakiviin. Tyypin I heterotsygootit eroavat toisistaan normaalien virtsan kystiinipitoisuuksien perusteella.
Toisin kuin tyypin I ja II homotsygooteilla, tyypin III homotsygooteilla esiintyy plasman kystiinipitoisuuden nousua oraalisen kystiinin annon jälkeen. Harris et al. raportoivat kystinurian genetiikan monimutkaisesta luonteesta mittaamalla virtsaan erittyvän kystiinin määrää kystinuriapotilaiden vanhemmilta (pakolliset heterotsygootit) ja havaitsivat täysin resessiivisiä alleeleja (molemmat vanhemmat erittivät kystiiniä viitealueella) ja dominoivia alleeleja (kumpikin vanhempi eritteli kystiiniä korkealla tasolla).
Kliinisen kystinurian luokittelemiseksi virtsan kystiini voidaan mitata koehenkilön kummaltakin vanhemmalta fenotyypiksi I (resessiivinen, virtsan kystiinipitoisuus < 100 µmol/g kreatiniinia), fenotyypiksi II (dominoiva, virtsan kystiinipitoisuus >1000 µmol/g kreatiniinia) ja fenotyypiksi III (osittain dominoiva, virtsan kystiinipitoisuus 100-1000 µmol/g kreatiniinia). Kystinuria voidaan luokitella myös sen perusteella, missä iässä oireet ilmenevät ensimmäisen kerran (ts. infantiili, juveniili, murrosikäinen).
Terveillä henkilöillä kystiinin erittymisen yläraja on 20 mg/g kreatiniinia (< 10 µmol/mmol kreatiniinia). Homotsygoottiset erittävät yli 400 mg/d (1,7 mmol/d), ja homotsygoottisten potilaiden kystiinin erittyminen on yleensä 600-1400 mg/d (2,5-5,8 mmol/d). Tyypin I ja III kystinuriaa sairastavat heterotsygootit erittävät alle 200 mg/d (0,8 mmol/d) eivätkä muodosta kiviä. Tyypin II heterotsygootit erittävät jopa 200-400 mg/d, mutta nämä potilaat voivat muodostaa kiviä. Kivien muodostumisen ilmaantuvuus lisääntyy, kun virtsan kystiinipitoisuus ylittää 700 µmol/l (170 mg/l).
Genetiikka
Viime vuosina molekyylibiologian kehittymisen myötä on kertynyt uutta tietoa kystinurian patofysiologiasta. Vuonna 1992 useat tutkijat raportoivat 2,3 kilobaarisen munuaisten cDNA:n (D2H tai rBAT) ekspressiokloonauksesta, joka indusoi kystiinin ja dibasisten aminohappojen natrium-riippumatonta ottoa cRNA:ta ruiskutetuissa Xenopus laevis -okosyyteissä. RBAT-geeni kartoitettiin kromosomille 2 (kaista 2p21) D2S119:n ja D2S288:n väliin. Genomitietokannassa tämä geeni on nyt nimetty SLC3A1:ksi.
Immunohistokemialliset ja in situ -hybridisaatiotutkimukset osoittivat, että rBAT ekspressoituu proksimaalisen tubuluksen ja ohutsuolen S3-segmentin (pars recta) soluissa luminaalisella harjasreunakalvolla. Vuonna 1995 Gasparini ja muut raportoivat, että SLC3A1:n mutaatioita esiintyi potilailla, joilla oli tyypin I kystinuria, mutta ei potilailla, joilla oli tyypin II tai III kystinuria. Tähän mennessä on kuvattu yli 160 erilaista mutaatiota, joihin kuuluu sekä pieniä että suuria DNA-emäsparien poistoja geenistä. Yksi SLC3A1:n yleisimmistä geneettisistä muutoksista on nimeltään M467T, ja useimmat mutaatiot ovat yleensä populaatiokohtaisia. M467T-mutaatio on melko yleinen Välimeren alueen väestöissä. Mielenkiintoista on, että sen osuus oli 40 prosenttia mutaatioista espanjalaisessa perhekohortissa, ja se oli harvinainen Kanadan Quebecissä tutkituilla potilailla.
Vuonna 1999 eristettiin SLC7A9-geeni (BAT1). Geeni koodaa 487 aminohappoa sisältävää proteiinia, ja se kartoitettiin kromosomille 19 (kaista 19q13) D19S414:n ja D19S220:n välille. BAT1-tuote näyttää olevan membraaniproteiini, jolla on 12 membraanin ylittävää aluetta. BAT1-geenin mutaatiot aiheuttavat todennäköisesti muun kuin tyypin I kystinurian (Rosenbergin tyypit II ja III). Mutaatiot 19q-lokuksessa ovat erityisen yleisiä libyalaisilla juutalaisilla, ja virtsakivitaudin riski potilailla, jotka perivät kaksi tällaista 19q-lokuksen mutaatiota, on suunnilleen verrattavissa riskiin potilailla, jotka perivät kaksi rBAT-mutaatiota.
116 mutaatiota tässä geenissä on raportoitu. Yleisin mutaatio libyalaisilla juutalaisilla johti siihen, että proteiinissa valiiniaminohappojäännös (V170M) korvattiin metioniinilla. Heterotsygooteilla, joilla on V170M-mutaatio, virtsan kystiinipitoisuudet vaihtelevat 86-1238 µmol/g kreatiniinia. Näin ollen osa V170M-heterotsygoottien arvoista vastaa tyypin III kystinuriaa ja osa tyypin II kystinuriaa.
Tyypin II ja tyypin III kystinurian ilmeinen erotteleva piirre on se, että tyypin II homotsygooteilla ei imeydy kystiiniä suolistosta. Vuonna 2000 Pras ehdotti uutta luokittelua, joka perustuu molekyylianalyysiin. Äskettäin Dello Strologo ja muut ovat ehdottaneet uutta geneettistä luokitusta seuraavasti:
-
Tyyppi A, SLC3A1:n molempien alleelien mutaatio: Heterotsygooteilla on normaali aminohappojen virtsaneritys.
-
Tyyppi B, SLC7A9:n molempien alleelien mutaatio: Heterotsygooteilla on tavallisesti lisääntynyttä virtsan erittyvää kystiini- ja dibaasisten aminohappojen erittymistä.
-
Tyyppi AB, kystinuria, joka johtuu 1 mutaatiosta SLC3A1:ssä ja 1 mutaatiosta SLC7A9:ssä: Sekatyyppinen kystinuria voi johtua kahden erilaisen mutanttigeenin yhteisvaikutuksesta, ja 19q-geenin koodaama proteiini on suorassa vuorovaikutuksessa rBAT:n kanssa proksimaalisen tubuluksen S3-segmentissä (ks. taulukko).
Martens ym. (2008) raportoivat hiljattain kolme geenideleetio-oireyhtymää, jotka liittyvät tyypin A kystinuriaan: 2p21-deleetio-oireyhtymä, hypotonia-kystinuria-oireyhtymä (HCS) ja hypotonia-kystinuria-oireyhtymän epätyypillinen muoto. SLC3A1:n ja PREPL:n molemmat alleelit puuttuvat HCS-potilailta. Epätyypillisessä HCS:ssä on lisäksi yksi geeni (C2orf34) poistettu.
Taulukko. Kystinurian luokittelu (Avaa taulukko uudessa ikkunassa)
Rosenberg ym.
Tyyppi I
Tyyppi II
Tyyppi III
Molekyylinen
Tyyppi I
Ei tyyppi I
Vastaava geeni
SLC3A1
SLC7A9
Band
2p21
19q13.1
Nro. mutaatioita
>60
Yleisin mutaatio
.
M467
V170M
Populaatio, johon mutaatio vaikuttaa
Välimerelliset espanjalaiset, 40 %
libyalaiset juutalaiset
Deletoitumisaste
.
54%
25%
Proteiini
rBAT
BAT1
Aminohappojen kuljetusjärjestelmä
Lokalisaatio proksimaalisessa muunnetussa tubuluksessa
S3
S1, S2
Transporterin ominaisuus
Korkea affiniteetti, alhainen kapasiteetti
Matalan affiniteetin, korkea kapasiteetti
Kliiniset piirteet
Homotsygootit
Oireet
.
noin 90 % oireilee
Heterotsygootit
Oireeton
noin 10 %-13% oireilevia
Virtsan kystiinipitoisuus
Normaali
normaalit +++++
normaalit +
Plasman kystiinipitoisuus suun kautta otetun kuormituskokeen jälkeen
Samat
Samat tai lievästi kohonneet
Nousseet
Suolikanavan kulkeutuminen
Puuttuu (kystiinin kulkeutumista ei tapahdu, lysiini), tai arginiinia)
Puuttuu
Vähenee
Uudet todisteet viittaavat siihen, että 4F2HC/4F2LC-kompleksi vastaa Y+L-aminohappojen kuljetusjärjestelmästä suolen ja munuaisten proksimaalisten tubulussolujen basolateraalisella pinnalla ja että 4F2LC-geenin (SLC7A7) mutaatiot kaistalla 14q11-13 aiheuttavat harvinaisen resessiivisen sairauden nimeltä lysiiniproteiini-intoleranssi.
Yhteenveto
rBAT, 90 kd:n kokoinen II-tyypin glykoproteiini, on jyrsijöiden proksimaalisten munuaistubulusten dibasisten aminohappojen korkea-affiniteettinen, natriumista riippumaton kuljettaja.Ihmisen rBAT-geeni on lokalisoitu kaistalle 2p21. Mielenkiintoista on, että linkitysanalyysi viittaa siihen, että tämä on sama alue, jolle on tunnistettu kystinurian lokus, SLC3A1.Maailmanlaajuisesti kystinuriapotilailla on tunnistettu 160 erilaista SLC3A1-geenin mutaatiota ja 116 SLC7A9-geenin mutaatiota.
Tyypin III ja II kystinuria (ei tyyppi I) on yhdistetty kaistaleeseen 19p13.1 (SLC7A9); SLC7A9-geenin tarkan roolin määrittämiseksi tarvitaan kuitenkin lisätutkimuksia. Noin 50 %:lle lapsista, joilla on kaksi SLC3A1-mutaatiota (klassinen homotsygoottinen tyypin I kystinuria), kehittyy vähintään yksi kivi ensimmäisen elinvuosikymmenen aikana.
 Kystiinin liukoisuus virtsaan.
Kystiinin liukoisuus virtsaan.