Renaal transport van cystine
Aminozuren worden gemakkelijk gefilterd door de glomerulus en ondergaan een bijna volledige reabsorptie door proximale tubulaire cellen. Slechts 0,4% van het gefilterde cystine komt in de urine terecht. Verschillende auteurs hebben aminozuurtransport bestudeerd in celmembranen van de proximale niertubuli van mensen, ratten en konijnen. Ten minste 2 transportsystemen zijn verantwoordelijk voor de reabsorptie van cystine, als volgt:
-
Hoge-affiniteitssysteem: Dit systeem is aangetast bij personen met cystinurie. Het hoge-affiniteitssysteem medieert de opname van 10% van L-cystine en de dibasische aminozuren aan het apicale membraan van het rechte derde segment (S3) van de proximale tubulus.
-
Lage-affiniteitssysteem: Dit systeem is aanwezig in het S1-S2 gedeelte van de proximale tubulus en is verantwoordelijk voor 90% van de L-cystine reabsorptie. Het proces met lage affiniteit versterkt het proces met hoge affiniteit. Na absorptie wordt elke molecule cystine intracellulair omgezet in 2 moleculen cysteïne. Cysteïne verlaat het basolaterale membraan.
Genetische studies van DNA van families met cystinurie onthullen een defect gen gelegen op chromosoom 2. Het gen dat codeert voor de cystine transporter, aanvankelijk rBAT genoemd, staat nu bekend als SLC3A1 (SLC voor solute carrier) in de internationale Genome Database. Een tweede cystinurie-gen op chromosoom 19 wordt SLC7A9 genoemd. Het normale SLC7A9-gen codeert voor een subeenheid van de cystine-transporter die b 0,+ AT (aminozuurtransporter) wordt genoemd. Het proces van cystine-opname wordt geactiveerd door de SLC3A1- en SLC7A9-genproducten. Het transport van L-cystine in het penseelgrensmembraanblaasje is natriumonafhankelijk en elektrogenetisch. Bij personen met cystinurie is de beweging van cystine of cysteïne vanuit de tubulaire cellen naar het bloed niet aangetast.
Intestinaal transport van cystine
De hoog-affiniteitstransporter is aanwezig in het apicale borstelgrensmembraan van het jejunum en is verantwoordelijk voor de absorptie van cystine en dibasische aminozuren. De meeste patiënten met cystinurie hebben een verminderde absorptie van cystine; cystine-deficiëntie is echter klinisch niet significant omdat de absorptie van korte-keten aminozuren niet wordt beïnvloed.
Normaal worden cystine en de andere dibasische aminozuren (d.w.z. ornithine, lysine, arginine) gefilterd bij de glomerulus en geherabsorbeerd in de proximale convolueerde tubulus door een hoog-affiniteit luminaal transmembraankanaal. Defecten in dit kanaal veroorzaken verhoogde niveaus van dibasische aminozuurafscheiding in de urine. Terwijl ornithine, lysine en arginine volledig oplosbaar zijn, is cystine relatief onoplosbaar bij een fysiologische urine-pH van 5-7, met een pKa-waarde van 8,3. Bij een urine pH-waarde van 7,8 en 8 is de respectieve oplosbaarheid van cystine bijna verdubbeld en verdrievoudigd.
Dent en Senior toonden aan dat de oplosbaarheid van cystine pH-afhankelijk is. De oplosbaarheid van cystine in urine is ongeveer 250 mg/L (1 mmol/L) tot een pH-waarde van 7, maar de oplosbaarheid neemt toe bij een hogere pH-waarde tot 500 mg/L (2 mmol/L) of meer boven een pH-waarde van 7,5, zoals weergegeven in de onderstaande afbeelding. Metingen in urine hebben duidelijk aangetoond dat de oplosbaarheid van cystine lineair toeneemt met toenemende ionensterkte. Pak en collega’s toonden aan dat ongeveer 70 mg extra cystine kan worden opgelost in elke liter oplossing, bij een toename van de ionensterkte van 0,005-0,3. Bovendien varieert de oplosbaarheid van cystine bij dezelfde ionensterkte en pH, afhankelijk van het specifieke type elektrolyt dat aanwezig is. Zie de afbeelding hieronder.
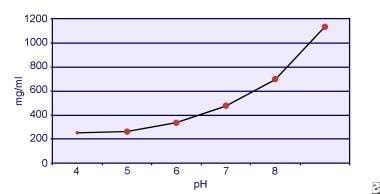 Cystine oplosbaarheid in urine.
Cystine oplosbaarheid in urine. In vitro-experimenten door Pak en Fuller in 1983 toonden aan dat de hoogste oplosbaarheid wordt bereikt in aanwezigheid van calciumchloride, gevolgd door magnesium- en natriumchloride. Verder wordt de oplosbaarheid van cystine ook beïnvloed door macromoleculen in de urine. Gebleken is dat de aanwezigheid van colloïden in normale urine de oplosbaarheid van cystine verhoogt; het mechanisme van deze werking is echter niet duidelijk. Omdat niets de kristallisatie van cystine remt, is de oververzadiging van de urine de belangrijkste determinant. Heterogene nucleatie van calciumoxalaat, brushiet, of hydroxyapatiet treedt niet op bij personen met cystinurie.
Risicofactoren voor cystinekristallisatie zijn (1) lage pH-waarde, (2) verlaagde ionensterkte, (3) de aanwezigheid van cystinekristallen, en (4) lage gehaltes van macromoleculen in de urine.
Cystine is een disulfidegebonden homodimer van cysteïne en heeft de volgende structuur:
COOH-CHNH2-CH2 -S-S-CH2-CHNH2- COOH Cystine
COOH-CHNH2-CH2 -SH Cysteine
Cystine wordt in de dunne darm opgenomen op een wijze die vergelijkbaar is met die van de nieren. Bij personen met cystinurie is de intestinale absorptie van cystine ook in meer of mindere mate verstoord. Metabolisme van methionine is een andere bron van serumcystine. Twee domeinen van membraanglycoproteïnen van type II zijn betrokken bij aminozuurtransport via het plasmamembraan. Het eerste is rBAT, en het tweede is 4F2HC (de zware keten van het 4F2 antigeen)
Twee derde van de personen met cystinurie die stenen vormen, maken zuivere cystine calculi, en een derde heeft een mengsel van cystine en calciumoxalaat calculi. In 2002 meldden Martins et al dat calciumoxalaat precipitatie optreedt door een salting-out proces, d.w.z. de vermindering in oplosbaarheid van een stof door de toevoeging van een andere stof aan het systeem, in plaats van door heterogene nucleatie. Hypocitraturie, hypercalciurie en hyperuricosurie worden ook vaak geassocieerd met cystinurie. Gezien hun relatief uniforme kristallijne structuur zonder gelamelleerde splijtvlakken, behoren zuivere cystinestenen tot de hardste op de steenfragiliteitsindex van Dretler.
Cystinurie is een autosomaal-recessieve ziekte die in 3 subtypes is onderverdeeld: Rosenberg I, II, en III. Cystinurie type I is de meest voorkomende variant en is in kaart gebracht op band 2p16.3. Type I heterozygoten vertonen normale aminoacidurie. Klassieke cystinurie, types II en III, werden verondersteld allelische varianten te zijn, maar koppelingsanalyses hebben uitgewezen dat type III een defect is van een ongekarakteriseerd gen (SLC7A9) op band 19q13.1. Heterozygoten van type II en III vertonen vaak cystinurie zonder cystine calculi en lopen mogelijk een verhoogd risico op andere vormen van urolithiasis. Type I heterozygoten onderscheiden zich door normale cystinegehalten in de urine.
In tegenstelling tot type I en type II homozygoten, vertonen type III homozygoten een toename van de cystineconcentratie in het plasma na orale toediening van cystine. Harris et al rapporteerden de complexe aard van de genetica van cystinurie door het niveau van de uitscheiding van cystine in de urine te meten bij de ouders (obligate heterozygoten) van probandes met cystinurie en vonden volledig recessieve allelen (beide ouders scheidden cystine uit in het referentiebereik) en dominante allelen (beide ouders scheidden cystine uit op hoge niveaus).
Om cystinurie klinisch te classificeren, kan het cystinegehalte in de urine bij elke ouder van een proband worden gemeten als fenotype I (recessief, cystinegehalte in de urine < 100 µmol/g creatinine), fenotype II (dominant, cystinegehalte in de urine >1000 µmol/g creatinine), en fenotype III (gedeeltelijk dominant, cystinegehalte in de urine 100-1000 µmol/g creatinine). Cystinurie kan ook worden ingedeeld op basis van de leeftijd waarop de symptomen voor het eerst optreden (d.w.z. infantiel, juveniel, adolescent).
In gezonde personen is de bovengrens voor cystine-uitscheiding 20 mg/g creatinine (< 10 µmol/mmol creatinine). Homozygoten scheiden meer dan 400 mg/d (1,7 mmol/d) uit, en de cystine-uitscheiding bij homozygote patiënten is meestal 600-1400 mg/d (2,5-5,8 mmol/d). Heterozygoten met cystinurie type I en III scheiden minder dan 200 mg/d (0,8 mmol/d) uit en vormen geen stenen. Type II heterozygoten scheiden tot 200-400 mg/d uit, maar deze patiënten kunnen wel stenen vormen. De incidentie van steenvorming neemt toe wanneer de cystineconcentratie in de urine hoger is dan 700 µmol/L (170 mg/L).
Genetica
In de afgelopen jaren, met de vooruitgang in de moleculaire biologie, zijn nieuwe inzichten geaccumuleerd met betrekking tot de pathofysiologie van cystinurie. In 1992 meldden verschillende onderzoekers de expressie klonering van een 2.3-kilobase renaal cDNA (D2H of rBAT) dat natrium-onafhankelijke opname van cystine en de dibasische aminozuren in cRNA-geïnjecteerde Xenopus laevis oöcyten induceerde. Het rBAT-gen werd in kaart gebracht op chromosoom 2 (band 2p21) tussen D2S119 en D2S288. Dit gen wordt nu SLC3A1 genoemd in de Genome Database.
Immunohistochemische en in situ hybridisatie studies toonden aan dat rBAT tot expressie komt in cellen van het S3 (pars recta) segment van de proximale tubulus en de dunne darm ter hoogte van het luminale borstelgrens membraan. In 1995 meldden Gasparini et al dat mutaties in SLC3A1 voorkwamen bij patiënten met type I cystinurie en niet bij patiënten met type II of III cystinurie. Tot op heden zijn er meer dan 160 verschillende mutaties beschreven, waaronder zowel kleine als grote deleties van DNA basenparen uit het gen. Een van de meest voorkomende genetische veranderingen in SLC3A1 heet M467T, en de meeste mutaties hebben de neiging populatie-specifiek te zijn. De M467T mutatie komt vrij vaak voor bij mediterrane populaties. Interessant is dat het 40% van de mutaties in een Spaans cohort van families uitmaakte en zeldzaam was bij patiënten bestudeerd in Quebec, Canada.
In 1999 werd het SLC7A9 (BAT1) gen geïsoleerd. Het gen codeert voor een eiwit van 487 aminozuren en werd in kaart gebracht op chromosoom 19 (band 19q13) tussen D19S414 en D19S220. Het BAT1-product blijkt een membraaneiwit te zijn met 12 membraan-overspannende gebieden. Mutaties in het BAT1-gen veroorzaken waarschijnlijk cystinurie zonder type I (Rosenberg type II en III). Mutaties op de 19q locus komen vooral voor bij Libische Joden, en het risico van urolithiasis bij patiënten die 2 van dergelijke 19q locus-mutaties erven is ongeveer vergelijkbaar met dat bij patiënten die 2 rBAT-mutaties erven.
116 mutaties in dit gen zijn gerapporteerd. De meest voorkomende mutatie bij Libische Joden resulteerde in een methionine ter vervanging van het valine aminozuur residu (V170M) in het eiwit. Bij heterozygoten met de V170M-mutatie varieert de cystineconcentratie in de urine van 86-1238 µmol/g creatinine. Sommige waarden bij V170M heterozygoten komen dus overeen met cystinurie type III en andere met cystinurie type II.
Een duidelijk onderscheidend kenmerk tussen cystinurie type II en type III is het ontbreken van intestinale cystine-absorptie bij homozygoten van type II. In 2000 stelde Pras een nieuwe classificatie voor op basis van moleculaire analyse. Onlangs hebben Dello Strologo et al. een nieuwe genetische classificatie voorgesteld, als volgt:
-
Type A, mutatie van beide allelen van SLC3A1: Heterozygoten vertonen een normaal aminozuur urinepatroon.
-
Type B, mutatie van beide allelen van SLC7A9: Heterozygoten vertonen meestal een toename van cystine en dibasische aminozuren in de urinaire uitscheiding.
-
Type AB, cystinurie veroorzaakt door 1 mutatie in SLC3A1 en 1 mutatie in SLC7A9: Gemengd-type cystinurie kan worden veroorzaakt door de interactie van 2 verschillende mutantgenen, en het eiwit dat wordt gecodeerd door het 19q-gen staat in directe interactie met rBAT in het S3-segment van de proximale tubulus (zie de tabel).
Martens et al. (2008) rapporteerden onlangs 3 gen-deletie syndromen geassocieerd met cystinurie type A: 2p21 deletie syndroom, hypotonie-cystinurie syndroom (HCS), en een atypische vorm van hypotonie-cystinurie syndroom. Beide allelen van SLC3A1 en PREPL ontbreken bij patiënten met HCS. Een extra gen (C2orf34) is verwijderd bij atypische HCS.
Tabel. Classificatie van Cystinurie (Open Tabel in een nieuw venster)
|
Rosenberg et al |
Type I |
Type II |
Type III |
|
Moleculair |
Type I |
Niet-Type I |
|
|
Verantwoordelijk gen |
SLC3A1 |
SLC7A9 |
|
|
Band |
2p21 |
19q13.1 |
|
|
Nr. mutaties |
>60 |
||
|
Meest voorkomende mutatie |
M467 |
V170M |
|
|
Populatie getroffen |
Mediterrane Spaanse personen, 40% |
Libische joden |
|
|
Verwijderingspercentage |
54% |
25% |
|
|
Proteïne |
rBAT |
BAT1 |
|
|
Amino zuur transport systeem |
|||
|
Lokalisatie in proximale omgezette tubule |
S3 |
S1, S2 |
|
|
Transporter kenmerk |
Hoge affiniteit, lage capaciteit |
Lage affiniteit, hoge capaciteit |
|
|
Klinische kenmerken |
|||
|
Homozygoten |
Symptomatisch |
ongeveer 90% symptomatisch |
|
|
Heterozygoten |
Asymptomatisch |
ongeveer 10%-13% symptomatisch |
|
|
Cystinegehalte in de urine |
Normaal |
Getaleerd +++++ |
Getaleerd + |
|
Cystinegehalte in het plasma na een orale belastingstest |
Zelfde |
Zelfde of lichte stijging |
Gestijgd |
|
Intestinaal transport |
Absent (geen transport van cystine, lysine, of arginine) |
Absent |
Reduceerd |
Nieuw bewijs suggereert dat het 4F2HC/4F2LC complex verantwoordelijk is verantwoordelijk is voor het Y+L aminozuurtransportsysteem aan het basolaterale oppervlak van darm- en nierproximale tubulaire cellen en dat de mutaties van het 4F2LC-gen (SLC7A7) op band 14q11-13 de zeldzame recessieve ziekte veroorzaken die lysine-eiwitintolerantie wordt genoemd.
Samenvatting
rBAT, een 90-kd type II glycoproteïne, is een hoog-affiniteit, natrium-onafhankelijke transporter voor dibasische aminozuren in de proximale convoluted renale tubuli in knaagdieren.Het humane rBAT gen is gelokaliseerd op band 2p21. Interessant genoeg suggereert koppelingsanalyse dat dit dezelfde regio is als waar een cystinurische locus, SLC3A1, is geïdentificeerd.Er zijn wereldwijd 160 verschillende mutaties in het SLC3A1-gen en 116 in het SLC7A9-gen geïdentificeerd bij patiënten met cystinurie.
Type III en II cystinurie (niet-type I) zijn in verband gebracht met band 19p13.1 (SLC7A9); verdere studies zijn echter nodig om de exacte rol van het SLC7A9-gen te bepalen. Ongeveer 50% van de kinderen met 2 SLC3A1-mutaties (klassieke homozygote cystinurie type I) ontwikkelt ten minste één steen binnen het eerste decennium van het leven.