Renal transport of cystine
Aminokwasy są łatwo filtrowane przez kłębuszek nerkowy i ulegają prawie całkowitej reabsorpcji przez komórki kanalików proksymalnych. Tylko 0,4% przefiltrowanej cystyny pojawia się w moczu. Różni autorzy badali transport aminokwasów w błonach komórkowych uzyskanych z kanalika bliższego nerki u ludzi, szczurów i królików. Co najmniej 2 systemy transportowe są odpowiedzialne za reabsorpcję cystyny, jak następuje:
-
System wysokiego powinowactwa: System ten jest zaburzony u osób z cystynurią. Układ wysokiego powinowactwa pośredniczy w wychwycie 10% L-cystyny i aminokwasów dibazowych w błonie apikalnej prostego trzeciego segmentu (S3) kanalika bliższego.
-
Układ niskiego powinowactwa: System ten jest obecny w części S1-S2 kanalika proksymalnego i odpowiada za 90% reabsorpcji L-cystyny. Proces niskiego powinowactwa wspomaga proces wysokiego powinowactwa. Po wchłonięciu, każda cząsteczka cystyny jest wewnątrzkomórkowo przekształcana do 2 cząsteczek cysteiny. Cysteina wydostaje się na zewnątrz przy błonie podstawnej.
Badania genetyczne DNA z rodzin z cystynurią ujawniają wadliwy gen zlokalizowany na chromosomie 2. Gen, który koduje transporter cystyny, początkowo określany jako rBAT, jest obecnie znany jako SLC3A1 (SLC od solute carrier) w międzynarodowej bazie danych Genome Database. Drugi gen cystynurii na chromosomie 19 nosi nazwę SLC7A9. Prawidłowy gen SLC7A9 koduje podjednostkę transportera cystyny zwanego b 0,+ AT (transporter aminokwasów). Proces wychwytu cystyny jest aktywowany przez produkty genów SLC3A1 i SLC7A9. Transport L-cystyny w pęcherzykach błony granicznej szczoteczek jest niezależny od sodu i elektrogenny. U osób z cystynurią przemieszczanie cystyny lub cysteiny z komórek kanalików do krwi nie jest zaburzone.
Jelitowy transport cystyny
Transporter o wysokim powinowactwie jest obecny w apikalnej błonie granicznej szczoteczek jelita czczego i jest odpowiedzialny za wchłanianie cystyny i aminokwasów dibazowych. Większość pacjentów z cystynurią ma upośledzone wchłanianie cystyny; jednak niedobór cystyny nie ma znaczenia klinicznego, ponieważ wchłanianie krótkołańcuchowych aminokwasów nie jest zaburzone.
Normalnie cystyna i inne aminokwasy dibazowe (tj. ornityna, lizyna, arginina) są filtrowane w kłębuszku i ponownie wchłaniane w kanaliku proksymalnym przez luminalny kanał transmembranowy o wysokim powinowactwie. Defekty w tym kanale powodują podwyższony poziom wydzielania aminokwasów dwuzasadowych w moczu. Podczas gdy ornityna, lizyna i arginina są całkowicie rozpuszczalne, cystyna jest stosunkowo nierozpuszczalna przy fizjologicznym poziomie pH moczu 5-7, z poziomem pKa wynoszącym 8,3. Przy poziomie pH moczu 7,8 i 8, odpowiednia rozpuszczalność cystyny jest prawie podwojona i potrojona.
Dent i Senior wykazali, że rozpuszczalność cystyny jest zależna od pH. Rozpuszczalność cystyny w moczu wynosi około 250 mg/L (1 mmol/L) do poziomu pH 7, ale rozpuszczalność wzrasta wraz z wyższym poziomem pH aż do 500 mg/L (2 mmol/L) lub więcej powyżej poziomu pH 7,5, jak przedstawiono na poniższym obrazku. Pomiary w moczu wyraźnie wykazały, że rozpuszczalność cystyny wzrasta liniowo wraz ze wzrostem siły jonowej. Pak i współpracownicy wykazali, że około 70 mg dodatkowej cystyny może być rozpuszczone w każdym litrze roztworu, przy wzroście siły jonowej z 0,005-0,3. Ponadto, przy tej samej sile jonowej i pH, rozpuszczalność cystyny różni się w zależności od konkretnego rodzaju elektrolitu. Patrz obrazek poniżej.
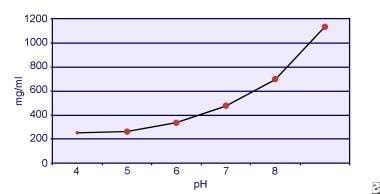 Rozpuszczalność cystyny w moczu.
Rozpuszczalność cystyny w moczu. Eksperymenty in vitro przeprowadzone przez Pak i Fuller w 1983 roku wykazały, że największa rozpuszczalność jest osiągana w obecności chlorku wapnia, a następnie chlorku magnezu i sodu. Ponadto, na rozpuszczalność cystyny mają również wpływ makromolekuły moczu. Wykazano, że obecność koloidu w normalnym moczu zwiększa rozpuszczalność cystyny; jednak mechanizm tego działania nie jest jasny. Ponieważ nic nie hamuje krystalizacji cystyny, głównym czynnikiem determinującym jest przesycenie moczu. Niejednorodne zarodkowanie szczawianu wapnia, szczoteczki lub hydroksyapatytu nie występuje u osób z cystynurią.
Do czynników ryzyka krystalizacji cystyny należą (1) niski poziom pH, (2) obniżona siła jonowa, (3) obecność kryształów cystyny i (4) niski poziom makrocząsteczek w moczu.
Cystyna jest homodimerem cysteiny połączonym disulfidem i ma następującą strukturę:
COOH-CHNH2-CH2 -S-S-CH2-CHNH2- COOH Cystyna
COOH-CHNH2-CH2 -SH Cysteina
Cystyna jest wchłaniana w jelicie cienkim w sposób podobny do wchłaniania przez nerki. U osób z cystynurią wchłanianie jelitowe cystyny jest również w różnym stopniu upośledzone. Metabolizm metioniny jest kolejnym źródłem cystyny w surowicy. Dwie domeny glikoprotein błonowych typu II zostały zaangażowane w transport aminokwasów przez błonę plazmatyczną. Pierwszą z nich jest rBAT, a drugą 4F2HC (łańcuch ciężki antygenu 4F2)
Dwie trzecie osób z cystynurią, u których tworzą się kamienie, tworzy kamienie czysto cystynowe, a jedna trzecia ma kamienie będące mieszaniną cystyny i szczawianu wapnia. W 2002 r. Martins i wsp. stwierdzili, że wytrącanie szczawianu wapnia następuje w wyniku procesu solenia, czyli zmniejszenia rozpuszczalności substancji w wyniku dodania innej substancji do układu, a nie w wyniku procesu heterogenicznego zarodkowania. Hipocitraturia, hiperkalciuria i hiperurykozuria są również często związane z cystynurią. Biorąc pod uwagę stosunkowo jednolitą strukturę krystaliczną bez lamelarnych płaszczyzn rozszczepienia, kamienie czysto cystynowe należą do najtwardszych w indeksie kruchości kamieni Dretlera.
Cystynuria jest chorobą dziedziczoną autosomalnie recesywnie, podzieloną na 3 podtypy: Rosenberg I, II i III. Cystynuria typu I jest najczęstszym wariantem i została zmapowana do pasma 2p16.3. Heterozygoty typu I wykazują prawidłową aminoacydurię. Klasyczna cystynuria, typy II i III, były uważane za warianty alleliczne, ale analizy powiązań ujawniły, że typ III jest defektem niescharakteryzowanego genu (SLC7A9) w paśmie 19q13.1. Heterozygoty typu II i III często wykazują cystynurię bez kamieni cystynowych i mogą być narażone na zwiększone ryzyko innych typów kamicy moczowej. W przeciwieństwie do homozygot typu I i typu II, homozygoty typu III wykazują wzrost stężenia cystyny w osoczu po doustnym podaniu cystyny. Harris i wsp. opisali złożoną naturę genetyki cystynurii, mierząc poziom wydalania cystyny z moczem u rodziców (heterozygot obligatoryjnych) probandów z cystynurią i stwierdzili allele w pełni recesywne (oboje rodzice wydalali cystynę w zakresie referencyjnym) oraz allele dominujące (oboje rodzice wydalali cystynę na wysokim poziomie).
Aby sklasyfikować cystynurię klinicznie, cystynę w moczu można zmierzyć u każdego z rodziców probanta jako fenotyp I (recesywny, poziom cystyny w moczu < 100 µmol/g kreatyniny), fenotyp II (dominujący, poziom cystyny w moczu >1000 µmol/g kreatyniny) i fenotyp III (częściowo dominujący, poziom cystyny w moczu 100-1000 µmol/g kreatyniny). Cystynuria może być również klasyfikowana na podstawie wieku, w którym po raz pierwszy pojawiają się objawy (tj. niemowlęcy, młodzieńczy, młodzieńczy).
U zdrowych osób górna granica wydalania cystyny wynosi 20 mg/g kreatyniny (< 10 µmol/mmol kreatyniny). Homozygoty wydalają więcej niż 400 mg/d (1,7 mmol/d), a wydalanie cystyny u pacjentów homozygotycznych wynosi zwykle 600-1400 mg/d (2,5-5,8 mmol/d). Heterozygoty z cystynurią typu I i III wydalają mniej niż 200 mg/d (0,8 mmol/d) i nie tworzą kamieni. Heterozygoty typu II wydalają do 200-400 mg/d, ale u tych pacjentów mogą tworzyć się kamienie. Częstość tworzenia się kamieni wzrasta, gdy stężenie cystyny w moczu przekracza 700 µmol/l (170 mg/l).
Genetyka
W ostatnich latach, wraz z postępem w biologii molekularnej, zgromadzono nowe spostrzeżenia dotyczące patofizjologii cystynurii. W 1992 roku, kilku badaczy zgłosiło klonowanie ekspresji 2,3-kilobazowego nerkowego cDNA (D2H lub rBAT), który indukował niezależny od sodu wychwyt cystyny i aminokwasów dibazowych w oocytach Xenopus laevis wstrzykniętych w cRNA. Gen rBAT został zmapowany na chromosomie 2 (pasmo 2p21) pomiędzy D2S119 i D2S288. Gen ten jest obecnie nazwany SLC3A1 w Genome Database.
Badania immunohistochemiczne i hybrydyzacja in situ ujawniły, że rBAT ulega ekspresji w komórkach segmentu S3 (pars recta) cewki proksymalnej i jelita cienkiego w luminalnej błonie granicznej szczotki. W 1995 roku Gasparini i wsp. donieśli, że mutacje w SLC3A1 występują u pacjentów z cystynurią typu I, a nie u pacjentów z cystynurią typu II lub III. Do chwili obecnej opisano ponad 160 różnych mutacji, w tym zarówno małe, jak i duże delecje par zasad DNA z tego genu. Jedna z najczęstszych mutacji genetycznych w SLC3A1 nosi nazwę M467T, a większość mutacji ma tendencję do bycia specyficznymi dla danej populacji. Mutacja M467T jest dość powszechna w populacjach śródziemnomorskich. Co ciekawe, stanowiła ona 40% mutacji w hiszpańskiej kohorcie rodzin i była rzadka u pacjentów badanych w Quebecu w Kanadzie.
W 1999 roku wyizolowano gen SLC7A9 (BAT1). Gen ten koduje 487-aminokwasowe białko i został zmapowany na chromosomie 19 (pasmo 19q13) pomiędzy D19S414 i D19S220. Produkt BAT1 wydaje się być białkiem błonowym z 12 regionami rozprzestrzeniającymi się w błonie. Mutacje w genie BAT1 prawdopodobnie powodują cystynurię niebędącą typem I (Rosenberg typ II i III). Mutacje w locus 19q są szczególnie częste wśród Żydów libijskich, a ryzyko kamicy moczowej u pacjentów, którzy odziedziczyli 2 takie mutacje w locus 19q jest mniej więcej porównywalne z ryzykiem u pacjentów, którzy odziedziczyli 2 mutacje rBAT.
116 mutacji w tym genie zostało zgłoszonych. Najczęstsza mutacja u Żydów libijskich powodowała zastąpienie reszty walinowej aminokwasem metioniny (V170M) w białku. U heterozygot z mutacją V170M stężenie cystyny w moczu waha się od 86-1238 µmol/g kreatyniny. Tak więc niektóre z wartości u heterozygot z mutacją V170M są zgodne z cystynurią typu III, a inne z cystynurią typu II.
Najwyraźniej cechą odróżniającą cystynurię typu II od cystynurii typu III jest brak jelitowej absorpcji cystyny u homozygot typu II. W 2000 roku Pras zaproponował nową klasyfikację opartą na analizie molekularnej. Ostatnio Dello Strologo i wsp. zaproponowali nową klasyfikację genetyczną, która przedstawia się następująco:
-
Typ A, mutacja obu alleli SLC3A1: Heterozygoty wykazują prawidłowy wzorzec wydalania aminokwasów z moczem.
-
Typ B, mutacja obu alleli SLC7A9: Heterozygoty zwykle wykazują wzrost wydalania cystyny i aminokwasów dibazowych z moczem.
-
Typ AB, cystynuria spowodowana 1 mutacją w SLC3A1 i 1 mutacją w SLC7A9: cystynuria typu mieszanego może być spowodowana interakcją 2 różnych zmutowanych genów, a białko kodowane przez gen 19q bezpośrednio oddziałuje z rBAT w segmencie S3 kanalika proksymalnego (patrz Tabela).
Martens i wsp. (2008) opisali ostatnio 3 zespoły delecji genów związane z cystynurią typu A: zespół delecji 2p21, zespół hipotonii-cystynurii (HCS) oraz atypową postać zespołu hipotonii-cystynurii. U pacjentów z HCS brak jest obu alleli SLC3A1 i PREPL. Dodatkowy gen (C2orf34) jest usunięty w atypowej postaci HCS.
Tabela. Klasyfikacja cystynurii (Otwórz tabelę w nowym oknie)
|
Rosenberg i wsp |
Typ I |
Typ II |
Typ III |
|
Molekularne |
Typ I |
Non-Typ I |
|
|
Gen odpowiedzialny |
SLC3A1 |
SLC7A9 |
|
|
Pasmo |
2p21 |
19q13.1 |
|
|
Nr. mutacji |
>60 |
||
|
Najczęstsza mutacja |
M467 |
V170M |
|
|
Populacja dotknięta mutacją |
Hiszpanie z basenu Morza Śródziemnego, 40% |
Żydzi libijscy |
|
|
Częstotliwość delecji |
54% |
25% |
|
|
białko |
rBAT |
BAT1 |
|
|
System transportu aminokwasów |
|||
|
Lokalizacja w kanaliku bliższym przekształconym |
S3 |
S1, S2 |
|
|
Charakterystyka transportera |
Wysokie powinowactwo, niska pojemność |
Niskie powinowactwo, wysoka pojemność |
|
|
Cechy kliniczne |
|||
|
Homozygoty |
Objawowe |
około 90% symptomatyczne |
|
|
Heterozygoty |
Asymptomatyczne |
około 10%-…13% objawowych |
|
|
Stopień cystyny w moczu |
Normalny |
Podwyższony +++++ |
Podwyższony + |
|
Stopień cystyny w osoczu krwi po doustnym teście obciążeniowym |
Taki sam |
Taki sam lub nieznacznie podwyższony |
Podwyższony |
|
Transport jelitowy |
Absent (brak transportu cystyny, lizyny, lub argininy) |
Absent |
Redukowany |
Ostatnie dowody sugerują, że kompleks 4F2HC/4F2LC odpowiada za za system transportu aminokwasów Y+L na powierzchni podstawnej komórek cewek proksymalnych jelit i nerek oraz że mutacje genu 4F2LC (SLC7A7) w paśmie 14q11-13 powodują rzadką chorobę recesywną zwaną nietolerancją białek lizyny.
Podsumowanie
rBAT, 90-kd glikoproteina typu II, jest transporterem o wysokim powinowactwie, niezależnym od sodu dla aminokwasów dibazowych w proksymalnych kanalikach nerkowych u gryzoni.Ludzki gen rBAT został zlokalizowany na paśmie 2p21. Co ciekawe, analiza powiązań sugeruje, że jest to ten sam region, w którym zidentyfikowano locus cystynurii, SLC3A1.U pacjentów z cystynurią na całym świecie zidentyfikowano 160 różnych mutacji w genie SLC3A1 i 116 w genie SLC7A9.
Cystynuria typu III i II (niebędąca typem I) została powiązana z pasmem 19p13.1 (SLC7A9); konieczne są jednak dalsze badania w celu określenia dokładnej roli genu SLC7A9. Około 50% dzieci z 2 mutacjami SLC3A1 (klasyczna homozygotyczna cystynuria typu I) rozwija co najmniej jeden kamień w ciągu pierwszej dekady życia.